An immortalized line of macrophages derived from BALB/c mice were used for the following protocol approved by the University of Guelph Animal Utilization Protocol 4193. Notably, other strains of mice or other sources of immortalized cells can be applied to the outlined protocol with sufficient testing to optimize the detailed parameters. The following protocol will navigate the steps beginning with a frozen vial of macrophage cells. Cells are stored in 10% FBS (fetal bovine serum), 1% L-glutamine and 5% Pen/Strep (Penicillin-Streptomycin) mixture to DMEM (Dulbecco's Modified Eagle Medium) and 20% DMSO (dimethyl sulfoxide).
1. Culturing of C. neoformans
- Using a glycerol stock, streak wildtype C. neoformans strain (H99) onto Yeast-extract peptone dextrose (YPD) agar plate to isolate single colonies.
- Incubate overnight for 16 h at 37 °C in a static incubator.
- Select a single colony of wildtype C. neoformans strain and culture in 5 mL of YPD broth in a loosely capped 10 mL test tube. Perform in quadruplicate.
- Incubate overnight for 16 h at 37 °C in a shaking incubator at 200 rpm.
- The following day subculture each overnight culture in a 1:100 dilution into 3 mL YPD broth.
- Measure Optical density (OD600nm) values of fungal culture to determine mid-log phase. Depending on the spectrophotometer, C. neoformans wildtype strain reaches mid-log phase commonly following 6 to 8 h incubation in YPD with general OD600nm values ranging 1.0 to 1.5.
- Once cells reach mid-log phase, take a 10 µL aliquot of fungal cell suspension into a clean 1.5 mL microcentrifuge tube and dilute 1:100 in sterile 1x phosphate buffered saline (PBS). Count the number of cells using a hemocytometer.
2. Culturing of macrophage cells
NOTE: Ensure work environment is sterilized prior to cell culture work.
- Preparation of cell culture medium
- Antibiotic-supplemented medium: Add 10% FBS (fetal bovine serum), 1% L-glutamine and 5% Pen/Strep (Penicillin-Streptomycin) mixture to DMEM (Dulbecco's Modified Eagle Medium). Filter medium through a 0.2 µm complete filter system and store at 4 °C.
- Antibiotic-free medium: Follow identical protocol but omit Pen/Strep mixture.
- Seeding macrophage cells
NOTE: Antibiotic-supplemented medium (2.1.1.) should be warmed to 37°C prior to cell culture work.- Thaw vial of macrophage cells quickly in 37 °C bead bath.
- Wash cells of freezing solution by resuspending the cells in 1 mL antibiotic-supplemented medium.
NOTE: Extra precaution is required to ensure the cells do not lyse due to harsh pipetting. - Transfer resuspended cells to a sterile 15 mL tube.
- Pellet cells by centrifuging at 400 × g for 5 min at room temperature.
- Carefully remove supernatant with a serological pipette or vacuum aspirator.
- Gently resuspend pellet in 10 mL of antibiotic supplemented medium.
- Gently pipette resuspended cells into a 60 x 15 mm cell culture-treated dish.
- Incubate dish at 37 °C with 5% CO2 overnight.
- Initial passage of macrophage cells
NOTE: Frozen cell stocks will typically contain between 5 to 10 million cells per vial (approx. 1 mL). On the subsequent day, macrophage cells that have survived the seeding protocol will adhere to the bottom of the cell culture dish and will be ready for passaging.- Warm antibiotic-supplemented medium (step 2.1.1) to 37 °C prior to cell culture work. Ensure work environment is sterilized prior to cell culture work. Visualize cells using a light microscope to ensure cells are adhered and healthy.
- Remove cell culture medium from the dish either with a serological pipette or a vacuum aspirator.
- Gently add 5-7 mL of sterile room-temperature PBS (phosphate-buffered saline) to the 60 x 15 mm dish.
- Gently tilt the dish to wash adhered cells.
- Remove PBS using a serological pipette or vacuum aspirator.
- Add 1 mL of cold PBS (stored at 4 °C) to the cells, tilt dish to distribute PBS over all cells, and allow to sit at room temperature for 1 min.
- Release cells by gentle tapping of the dish, pipetting cold PBS against the cells, or by using a cell scraper.
- Add 9 mL of antibiotic-supplemented medium to the dish.
- In a new 60 x 15 mm dish, add 9 mL of fresh antibiotic-supplemented medium and 1 mL of resuspended cells from the original dish.
- Once the number of desired passaging plates have been filled, resuspended cells from the original dish can be discarded.
- Incubate the new dish at the settings mentioned above (step 2.2.8).
- Perform subsequent passaging when cells have reached 70-80% confluence (approximately every 2 days depending on the cell line and medium used).
NOTE: Macrophage cells should be passaged a minimum of five times prior to infection experiments. Depending on the cell line, infection experiments should be performed by 25 to 30 passages. After 25 to 30 passages, cells should be frozen or discarded. Section 2.4 or 2.5 can be chosen based on the experimental plans. Section 2.4 will be used for the following sections.
- Seeding macrophage cells prior to infection
- Perform passaging protocol steps 2.3.1 to 2.3.7.
- Using a hemocytometer or automated cell counter determine the cell density (cells/mL).
- Transfer 0.3 x 106 macrophage cells into a single well of a 6-well cell culture plate.
- Adjust total volume of well to 1 mL using antibiotic-supplemented medium (step 2.1.1).
- Repeat steps 2.4.3 and 2.4.4 until 8 wells have been filled (4 wells filled in two separate plates).
- Optionally, fill the remaining two wells with 1 mL of room-temperature PBS to maintain moisture levels during incubation.
- Allow cells to grow under incubation conditions mentioned in step 2.2.8 for 2 d prior to infection.
- Seeding macrophage cells on the day of infection
- Perform passaging protocol steps 2.3.1 to 2.3.7.
- Using a hemocytometer or automated cell counter determine the cell density (cells/mL).
- Seed 1.2 x 106 macrophage cells into a single well of a 6-well cell culture plate.
- Adjust total volume of well to 1 mL using antibiotic-supplemented medium (2.1.1).
- Repeat 2.4.3 and 2.4.4 until 8 wells have been filled (4 wells filled in two separate plates).
- Optionally, fill the remaining two wells with 1 mL of room-temperature PBS to maintain moisture levels during incubation.
- Incubate cells under conditions mentioned in step 2.2.8 for 3 h to allow cells to adhere to wells.
3. Infection of macrophage cells with C. neoformans
NOTE: Upon reaching 70-80% confluence, there will be approx. 1.2 x 106 macrophage cells per well. To achieve the desired multiplicity of infection (MOI) of 100:1, 1.2 x 108 fungal cells are required for each reaction. Cultures must be set accordingly in biological quadruplicate.
DISCLAIMER: A MOI of 100:1 has achieved desirable results in our research group and is meant as a suggestion to readers. A lower MOI may be required for more infectious C. neoformans strains or for less resilient macrophage cell lines. Verification of infection (section 3.5) can be used to determine the ideal MOI for particular C. neoformans – macrophage combinations.
- Preparation of fungal cells
- Follow step 1 for growth of C. neoformans to mid-log phase.
- Collect and centrifuge cells at 1,500 x g for 10 min, gently wash pellet with sterile room-temperature PBS, and repeat for a total of three washes.
- Resuspend cells in antibiotic-free cell culture medium (step 2.1.2) to achieve a concentration of 1.2 x 108 cells/mL.
- Preparation of macrophage cells
- Visualize each well in the 6-well plate to ensure that cells have reached 70-80% confluence. Alternatively, cells can be measured to achieve approx. 1.2 x 106 macrophage cells per well.
- Follow steps 2.3.1 to 2.3.4.
- Co-culture of C. neoformans and macrophage cells
- Add 1 mL of the resuspended C. neoformans cells (step 3.1.3) to 4 wells containing macrophage cells prepared in section 3.2.
NOTE: The number of plates required will need to be calculated prior to beginning the experiment. Add 1 mL of antibiotic-free medium (step 2.1.2) to empty wells. - Allow cells to incubate under conditions listed (step 2.2.8) for 3 h.
- Remove cell culture medium from the plate either with a serological pipette or a vacuum aspirator.
- Gently add 1 mL of sterile room-temperature PBS.
- Gently tilt the plate to wash non-attached or non-phagocytosed extracellular C. neoformans cells.
- Remove PBS using a serological pipette or vacuum aspirator, repeat for a total of three washes. Repeat 3.3.4 to 3.3.5 two more times.
- Add 1 mL of the resuspended C. neoformans cells (step 3.1.3) to 4 wells containing macrophage cells prepared in section 3.2.
- Uninfected macrophages
- Likewise, use 4 wells of a 6 well plate to serve act as macrophage-only samples. Add 1 mL of antibiotic-free medium (step 2.1.2) to these wells.
- Repeat steps 3.3.2 to 3.3.6.
- Verification of infection
NOTE: Using a cytotoxicity assay, infection proficiency can be measured. The following protocol will highlight application of a cytotoxicity product to measure LDH (lactate dehydrogenase) release. Other cytotoxicity products can also be used.- Preparation of C. neoformans infection of macrophage cells
- Repeats steps 1, 2, and 3 (up to 3.5). The LDH assay can be performed in triplicate, if preferred.
- Following step 3.3.6, add 1 mL of antibiotic-free medium (2.1.2) to each well in the 6-well plate.
- Repeat steps 2.4.3 and 2.4.4 until 3 wells plus 3n wells have been filled (where n is the number of time points measured).
- Incubate cells under conditions listed at step 2.2.8.
- At selected time points (e.g., 1, 3, 6, 12, and 24 hours) collect supernatant for measurement of LDH release according to manufacturer’s instructions
- At the same time points, uninfected macrophage cells will be lysed to determined value for maximum cytotoxicity.
- Calculate cytotoxicity as follows:
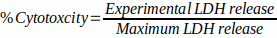
- Preparation of C. neoformans infection of macrophage cells
4. Sample collection
- Co-culture and uninfected macrophage collection
- Add 1 mL of cold PBS to the cells (from 3.3.6 and 3.4.2) and allow to sit at room temperature for 1 min.
- Release cells from the plate by gentle tapping of the plate or gentle pipetting cold PBS against the cells.
NOTE: Cell scraper should be avoided as this could cause lysis of cells. - Pipette resuspended cells into a 15 mL tube.
- Centrifuge cells at 400 x g for 5 min at room temperature, and remove the supernatant.
- Process cells (as detailed in step 5) immediately or flash frozen in liquid nitrogen and stored at -80 °C for later processing.
5. Cellular proteome
NOTE: Sufficient lysis must be optimized for the cell type analyzed (i.e., the quantity of cycles and amplitudes depends on cell pellet size and the power percentage of probe sonicator model).
- Infected macrophage cell lysis
- Resuspend pelleted cells (step 4.1.5) in 300 µL of 100 mM Tris-HCl (pH 8.5) consisting of a freshly dissolved protease inhibitor cocktail tablet.
NOTE: One protease inhibitor cocktail tablet is added to 10 mL of ice cold 100 mM Tris-HCl (pH 8.5) prior to beginning the experiment. - Probe sonicate cells in an ice bath for 15 cycles of 30 s on and 30 s off, to lyse the cells.
- Centrifuge cells briefly for 30 s at 400 x g, careful not to form a pellet, just to remove liquid on sides of tubes followed by transfer of sample to a 2 mL Lo-bind microcentrifuge tube.
- Add 1:10 volume of 20% SDS to a final concentration of 2%.
- Add 1:100 volume of 1 M dithiothreitol (DTT) to a final concentration of 10 mM and mix the sample thoroughly by pipetting, followed by incubation on a thermal heating block at 95 °C for 10 min at 800 rpm agitation. Next, cool to room temperature (cooling may be done on ice).
- Add 1:10 volume of 0.55 M iodoacetamide (IAA) to obtain a final concentration of 55 mM and mix the sample thoroughly by pipetting. Incubate at room temperature in the dark for 20 min.
- Add 100% acetone to obtain a final concentration of 80% acetone and store sample overnight at -20°C to precipitate proteins.
- Resuspend pelleted cells (step 4.1.5) in 300 µL of 100 mM Tris-HCl (pH 8.5) consisting of a freshly dissolved protease inhibitor cocktail tablet.
- Protein digestion
- The next day, collect the precipitate pellet by centrifugation for 10 min at 10,000 x g and 4 °C. Discard supernatant and wash pellet with 500 µL of 80% acetone. Repeat for a total of two washes. Air dry pellet at room temperature following washes.
- Resolubilize protein pellet in 100 µL of 8 M urea/40 mM HEPES, to ensure complete solubilization, vortex or sonicate in an ice water bath for 15 cycles of 30 s on and 30 s off.
NOTE: Adjustment of the volume of urea/HEPES is determined on size of precipitated cell pellet, if alterations occur all downstream volumes must be appropriately adjusted. - Quantify protein concentration using a protein assay (e.g., BCA protein assay) according to manufacturer’s instructions and adjust for background measurement by blank normalization with 8 M urea/40 mM HEPES.
- Add 300 µL of 50 mM ammonium bicarbonate to obtain a final concentration of 2 M urea.
NOTE: Opportunity to normalize protein concentration for downstream measurements, suggested to digest 100 µg of protein and store the remaining undigested sample by flash freezing in liquid nitrogen then store at -20 °C for short-term, or at -80 °C for longer-term. - Add 2:50 (v/w) enzyme-to-protein ratio of trypsin/Lys-C protease mixture on ice and gently tap tube to mix, incubate overnight at room temperature.
- Following incubation, stop digestion by adding 1:10 volume stopping solution (20% acetonitrile, 6% trifluoroacetic acid) and centrifuge samples at 10,000 x g for 5 min at room temperature.
- Collect supernatant (consists of digested peptides) and discard any pelleted debris or precipitate.
- Peptide desalting
- Activate a C18 Stop And Go Extraction (STAGE) tip (consisting of 3 layers C18 resin in a 200 µL pipette tip) by adding 100 µL of 100% acetonitrile and centrifuge at 1,000 x g for 2 min.
- Equilibrate the C18 STAGE tip by adding 50 µL of Buffer B (80% (v/v) acetonitrile, 0.5% (v/v) acetic acid) and centrifuge at 1,000 x g for 2 min.
- Equilibrate the C18 STAGE tip by adding 200 µL of Buffer A (2% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid, 0.5% (v/v) acetic acid) and centrifuge at 1,000 x g for 3-5 min.
- Add ~50 µg of digested sample onto C18 STAGE tip and centrifuge at 1,000 x g for 3-5 min, or until sample has passed through spin column. Flash freeze the remaining digested sample in liquid nitrogen and store at -20°C, until needed.
- Wash the C18 STAGE tip with 200 µL of Buffer A and centrifuge at 1,000 x g for 3-5 min.
- Add 50 µL Buffer B to the C18 STAGE tip and centrifuge at 500 x g for 2 min. Collect eluted peptides in 0.2 mL PCR tubes.
- Dry the eluted peptides in a vacuum centrifuge for 30-40 min at maximum speed. Completely dried samples may be stored at room temperature or at -20 °C until processed.
NOTE: Dried and desalted peptides are appropriate sample submissions to mass spectrometry facilities for processing and may be shipped at ambient temperature.
6. Mass spectrometry
- Reconstitute peptides in 10 µL Buffer A and measure concentration necessary to inject ~1.5 to 3 µg peptides onto the MS column. Amount of sample will depend on instrumentation.
- Use a pre-determined gradient of acetonitrile (approx. 5-60%) in 0.5% acetic acid over a desired time (e.g., 2 h) to separate peptides by high-performance liquid chromatography, followed by electrospray ionization into the mass spectrometer.
- Acquire MS scans using a high-resolution mass spectrometer in data dependent acquisition mode (m/z 300 to 1650).
NOTE: Gradient percentage and length are determined by the experiment and the user. Precise mass spectrometer settings depend on instrumentation, experiment, and user preference.
7. Data analysis
NOTE: MS data can be processed with numerous bioinformatics pipelines. In this protocol, we describe processing using the publicly available MaxQuant and Perseus platforms but recommend individual users to evaluate bioinformatic tools appropriate for the analysis, preference, and usage.
- Load unprocessed data files (directly from the MS instrument) using MaxQuant software. Identify proteins under the modified MaxQuant search parameters; minimum of two unique peptides necessary for protein identification using a target decoy approach for a false discovery rate of 1%, implement label-free quantification with matching between runs, incorporate the organisms FASTA file obtained from the UniProt database (i.e., Cryptococcus neoformans H99, Mus musculus) to identify and quantify present peptides with the Andromeda search engine. Consult public MaxQuant online tools for detailed tutorials (see Table of Materials).
- Upload the MaxQuant output file (‘proteingroups.txt’) into Perseus.
- Filter rows containing potential false positives and contaminants, as well as only modified by site peptides with the ‘Filter rows based on categorical column’.
- Transform data values on log2 scale.
- Create data set groups by providing categorical annotation to the rows.
- Filter dataset by valid values to define a cut-off for protein detection.
NOTE: For a stringent and robust analysis a >50% identification rate is suggested. For example, if four replicates were processed then a minimum number of three valid values would be selected. - If preferred, impute data by replacing missing values from the normal distribution.
NOTE: Imputed values are optimized based on normal distribution and provides a random LFQ intensity to replace ‘NaN’ placeholders to simulate typical abundance measurements. This imputation provides a platform for downstream statistical analysis that require quantifiable data. - Add annotations to the protein rows (e.g. protein names, Gene Ontology terms).
NOTE: This Perseus workflow generated is now a robust framework for further bioinformatic processing, statistical analysis, and data visualization, consult public Perseus online tools for detailed tutorials (see Table of Materials).
The protocol outlined above enables identification and quantification of proteins derived from both the fungal pathogen, C. neoformans, and the host, macrophage cells, in a single experiment. Following co-culture, cells are collected and processed together and bioinformatically separated based on peptide profiles specific to each species. This is a powerful approach for defining the interplay of the host-pathogen relationship during infection. The number of proteins identified from the experiment depends on the starting material, sample preparation, gradient length, MS instrumentation, and bioinformatic workflow. Using the protocol described herein, we typically, identify approx. 8,000 proteins from the experiment with 1,500 C. neoformans proteins and 6,500 host proteins. Following processing of the datasets, we generate a Principal Component Analysis (PCA) plot to observe critical factors driving our analysis (Figure 2A). Here, we observe the largest component of separation among the data is infected vs. non-infected samples, as we would anticipate from the experimental design (component 1, 79.8%), and a second distinguishing feature of the samples is biological variability (component 2, 5.7%). Next, a Pearson correlation combined with hierarchical clustering by Euclidean distance groups the samples and enables quantification of the variability among the replicates (Figure 2B). In our analysis, we observed distinct clustering of infected vs. non-infected samples and replicate reproducibility ranging from 95-96%, representing good reproducibility among the replicates. Lastly, we perform a Student’s t-test corrected for multiple hypothesis testing using a Benjamini-Hochberg false discovery rate (FDR) (p-value ≤ 0.05; FDR = 0.01; s0 = 1) to identify proteins with significant differences in abundance during infection compared to non-infected controls (Figure 2C). Here, we identify 760 proteins with significant changes in abundance, including 117 host proteins with 86 showing a significant decrease and 31 showing a significant increase upon infection. Notably, we also observe significant increases in abundance of fungal proteins, as expected during infection. With these data, subsequent analyses, including network mapping, in silico characterization, and follow-up experiments are performed to validate the data and explore the molecular mechanisms underpinning the host response to virulence.
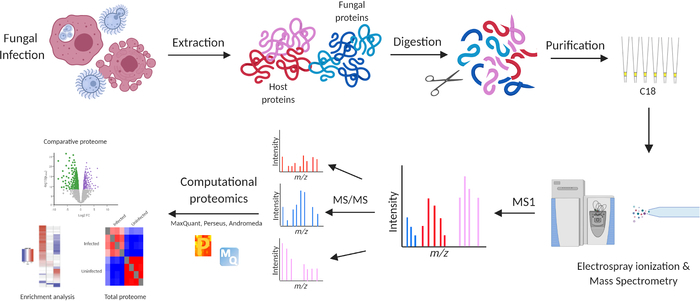
Figure 1: Mass spectrometry-based proteomics workflow for analysis of macrophages infected with C. neoformans. The workflow begins with collection of macrophages either infected with C. neoformans or non-infected controls. Proteins are extracted by mechanical and chemical disruption, followed by reduction and alkylation, acetone precipitation, and enzymatic digestion. Peptides are purified on C18 STAGE tips, separated by high-performance liquid chromatography, subjected to electrospray ionization, and measured on a high-resolution mass spectrometer. Data is processed, analyzed, and visualized in the publicly available bioinformatics platforms, MaxQuant (with Andromeda) and Perseus14,15,16. Experiments performed in biological quadruplicate. Please click here to view a larger version of this figure.
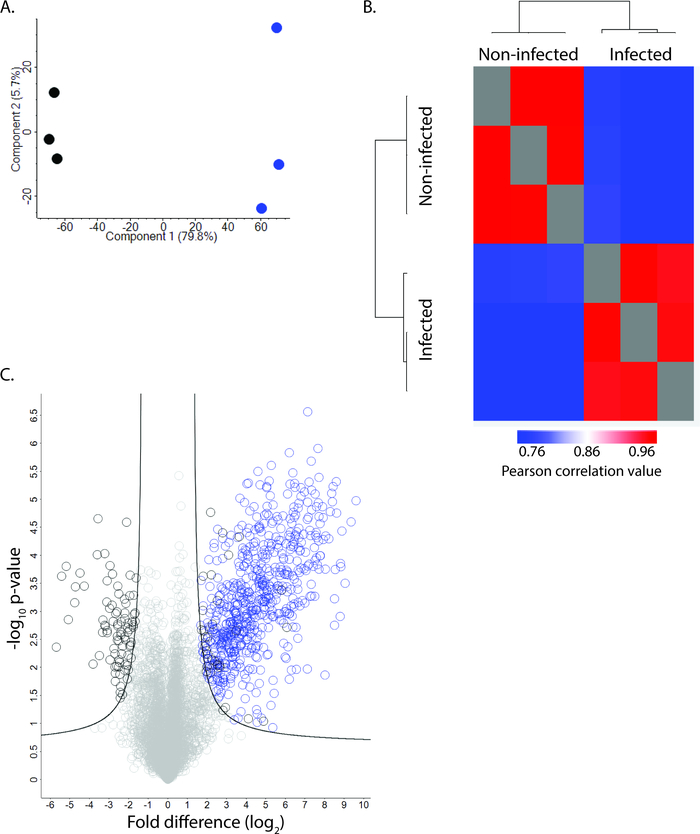
Figure 2: Representative data for C. neoformans infection of macrophage cells. (A) Principal component analysis demonstrates distinction between infected vs. non-infected macrophage (component 1, 79.8%), and clustering of biological replicates (component 2, 5.7%). (B) Heat map of Pearson correlation plotted by hierarchical clustering by Euclidean distance to show clustering of samples (infected vs. non-infected) and replicate reproducibility (>95%). (C) Volcano plot of identified proteins. Blue = fungal proteins with significant change in abundance; black = macrophage proteins with significant change in abundance. Student’s t-test (p-value ≤ 0.05), FDR = 0.01; s0 = 1. Please click here to view a larger version of this figure.
