This protocol to study glutamate receptor trafficking is based on differential labeling of receptors expressed at the cell surface and those expressed in internal membranes. Segregation is achieved by the labeling the receptors before and after membrane permeabilization, using the same primary antibody but a secondary antibody conjugated to a different fluorophore. As outlined by the optional steps included the protocol, this is a very versatile method for interrogating different receptor trafficking processes, such as internalization and recycling, and can be easily adapted to the investigator's needs (Figure 1).
First, a method is provided for the quantification of receptors expressed at the neuronal surface. This protocol allows for quantification of basal surface expression as well as studying the molecular mechanisms by which different drugs or mutations alter basal levels of surface-expressed receptors. Surface-expressed receptors were first labeled by incubating with both primary and secondary antibody prior to permeabilization. After permeabilization with 0.25% Triton X-100, the intracellular pool of receptors is accessible for antibody labeling. To illustrate this protocol, surface vs. intracellular staining of AMPA receptors (AMPARs) in control cultures and after induction of cLTP was conducted. Specifically, live cells were labeled using an antibody against an extracellular epitope in the GluA1 subunit of the AMPAR, and cells were fixed then labeled with Alexa 555-conjugated secondary. The cells were then permeabilized and labeled with same anti-AMPAR antibody and incubated with a secondary antibody conjugated to Alexa 647. This dual labeling allows clear visualization of the two GluA1 populations.
After acquiring confocal images, the fluorescent signal can be easily quantified to show an increase in surface expression, relative to the intracellular population, following cLTP (Figure 2A,B). As a control, the permeabilization step was skipped (incubation with 0.25% Triton X-100) in a sister coverslip and a primary antibody was used against PSD-95, a standard intracellular marker for excitatory synapses. As shown in Figure 2C, no signal for PSD-95 can be obtained in non-permeabilized cells, demonstrating the integrity of the plasma membrane. This indicates that the signal obtained for "surface GluA1" indeed corresponds to surface-expressed receptors (i.e., signal for surface-expressed GluA1 does not include intracellular receptors). Importantly, a minimal signal for "internalized GluA1" can be observed under non-permeabilization conditions, showing that all surface epitopes are occupied by the initial round of antibody labeling (i.e., signal for intracellular GluA1 does not included surface-expressed receptors).
A second process that can be examined utilizing this protocol is the internalization of surface-expressed receptors. Specifically, surface receptors are labeled on a live cell, and neurons are returned to the incubator for a given time to undergo receptor internalization by clathrin-mediated endocytosis. Following this step, cells are fixed to preserve the spatial expression of primary antibody-labeled receptors (i.e., surface-expressed and internalized receptors). Then, surface receptors (i.e., those which have not been internalized in the time period of interest), are labeled with a secondary antibody prior to permeabilization. Following permeabilization, receptors that have been internalized are labeled by a secondary antibody with a different fluorophore.
In this protocol, it was examined how a particular phosphorylation within the PDZ ligand of the GluN2B subunit of NMDARs (at S1480) induces NMDAR internalization. To do so, primary cultures were transfected with a phospho-mimetic receptor, in which serine (S1480) had been substituted by glutamate (E). The resultant mutant, GluN2B S1480E, acts as a "constitutively-phosphorylated" form of GluN2B. To ease labeling of GluN2B and identify the phospho-mimetic mutant, GFP was used as an epitope tag on the extracellular side of GluN2B (GFP-GluN2B S1480E). Surface receptors on live cells were labeled with an anti-GFP antibody for 15 min at RT. Next, the excess antibody was washed with conditioned media and returned the cells to 37 °C for 30 min to allow for endocytosis. Then, cells were fixed to freeze receptor movement. Receptors that remained on the surface were then labeled with Alexa 555-conjugated secondary antibody prior to permeabilization.
To identify receptors that had been internalized during the 30 min incubation period, cells were permeabilized, and the internalized receptors (already labeled with a primary antibody) were then labeled with Alexa 647-conjugated antibody. Again, this dual-labeling strategy allows quantification of the proportion of internalized receptors. This example highlights that GluN2B phosphorylation at S1480 promotes receptor internalization, as the phospho-mimetic mutant S1480E displayed a much higher internalization ratio compared to WT receptors (Figure 3A,B). As a control, a sister culture at 4 °C was maintained in conditioned media during internalization to strongly slow the process. As expected, no signal was obtained for "internalized" receptors under these conditions (Figure 3C).
Lastly, this protocol can be utilized to examine the recycling of previously internalized receptors. This protocol variation is a continuation of the internalization protocol, by following these receptors back to the cell surface. There are two crucial components to this variation. Firstly, receptors which remained stably expressed at the surface during the entire protocol (i.e., receptors that were not internalized or recycled) must be "blocked" so that they are not mistaken for recycled receptors. To do so, before performing the recycling step, live neurons are incubated with high concentrations of Fab that interact with primary antibody-labeled receptors expressed at the surface to prevent further binding of the fluorophore-conjugated secondary antibody. Second, internalization should be prevented during the recycling phase, so that recycled receptors are not repeat internalized. This was done by adding dynasore to the media during recycling as this drug blocks processes reliant on dynamin such as clathrin-mediated endocytosis, such as NMDAR internalization. In this protocol, studying the trafficking of the phospho-mimetic mutant GluN2B S1480E was performed as well as surface labeling of GFP-GluN2B on live cells, which allowed for internalization during a 30 min period as explained before.
Following this, surface receptors that were not internalized during this period were blocked by incubating the cells with Fab for 20 min at RT. Next, cells were again incubated at 37 °C for 45 min to allow for previously internalized GluN2B to be recycled back to the cell surface. Dynasore was present in the media during the recycling step. Fixation of the cells following this step allows for the identification of recycled receptors (i.e., those that are unblocked and expressed on the cell surface) and internalized receptors (i.e., those that are primary antibody labeled and intracellular). By 1) labeling recycled receptors with an Alexa 555-conjugated secondary antibody prior to permeabilization and 2) labeling internalized, but not recycled, receptors with Alexa 647-conjugated secondary antibody, a recycling ratio was generated to show that GluN2B S1480E does not have any effect on NMDAR recycling (Figure 4A,B). As a control, it was ensured that complete blocking of surface-expressed receptors occurred by incubating sister coverslips in the presence or absence of Fab, followed with PFA fixation. As shown in Figure 4C, a strong signal can be observed for surface-expressed GluN2B in the absence of Fab blocking. This signal disappears in Fab-treated cultures, demonstrating that the blocking protocol is sufficient to completely block the surface-expressed epitopes and that the surface signals observed after recycling indeed correspond to receptors trafficked back to the plasma membrane.
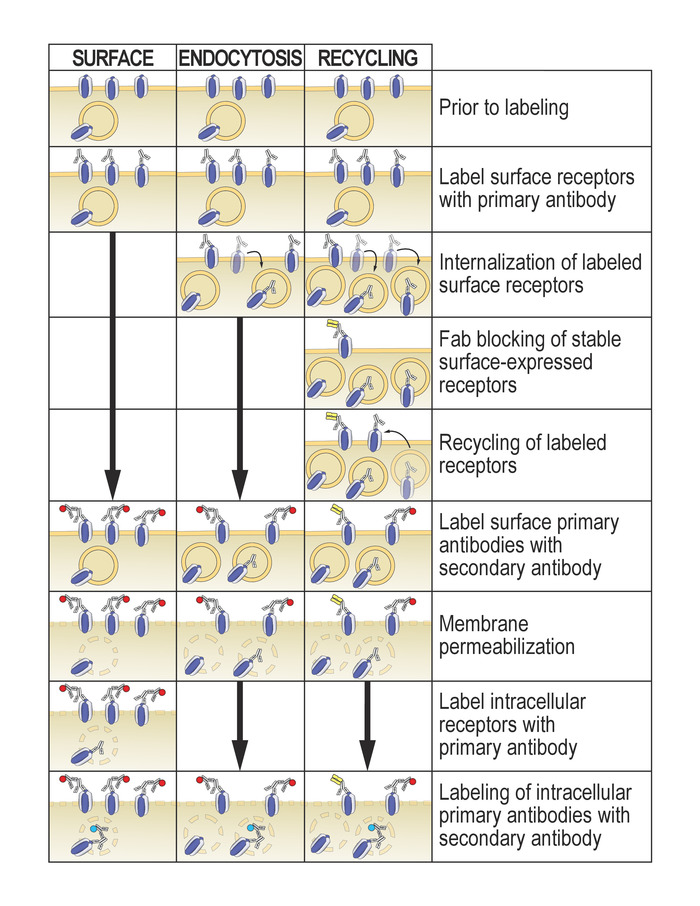
Figure 1: Schematic of protocol variations to study receptor surface expression, receptor internalization (endocytosis), and receptor recycling. Please click here to view a larger version of this figure.
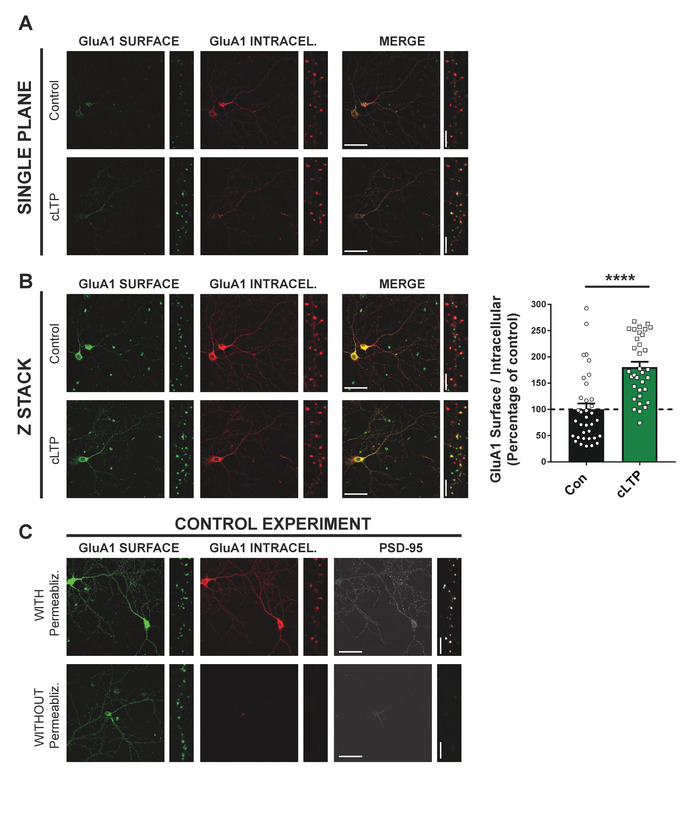
Figure 2: cLTP increases surface expression of GluA1. Primary hippocampal neurons at DIV21 were subjected to chemical LTP (cLTP) by incubating with glycine-containing ECS. Distinct labeling of surface-expressed (red) vs. intracellular (blue) GluA1 populations reveals the expected increase in surface expression of AMPAR. (A) Single plane and (B) Z-stacked (maximum intensity projection) confocal pictures. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Graph shows the increased surface expression of GluA1 after cLTP protocol. Surface expression index: surface/intracellular receptors (n = 3; number of cells: con = 7; cLTP = 7; values represent mean ± SEM; ****p < 0.0001 using Mann-Whitney U test). (C) Control experiment in which the permeabilization step was skipped. In addition to surface and internal GluA1, the intracellular excitatory synaptic marker PSD-95 was evaluated. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Please click here to view a larger version of this figure.
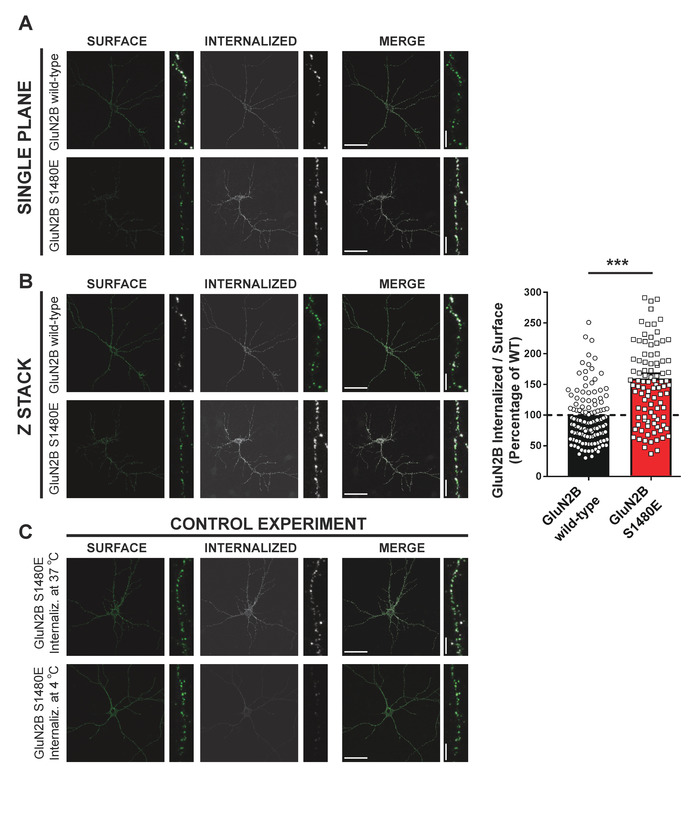
Figure 3: Phosphorylation of GluN2B at S1480 promotes NMDAR internalization. Primary hippocampal neurons were transfected with either GFP-GluN2B WT or the phospho-mimetic mutant GFP-GluN2B S1480E on DIV11-12. Following 3-4 days of protein expression, surface GFP was labeled on live cells with a rabbit anti-GFP antibody and cells were then returned to 37 °C to allow for receptor internalization by endocytosis. Surface-expressed exogenous receptors were visualized with Alexa 555-conjugated secondary antibody, and the internalized population identified after permeabilization using Alexa 647-conjugated antibody. For clarity, surface GFP-GluN2B is pseudocolored in green and internalized GFP-GluN2B is pseudocolored in white. (A) Single plane and (B) Z-stacked (maximum intensity projection) confocal pictures. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Graph shows the elevated internalization displayed by the phospho-mimetic mutant GluN2B S1480E. Internalization index: internalized receptors/surface-expressed receptors (n = 6; number of cells: WT = 34; S1480E = 28; values represent mean ± SEM; ***p < 0.001 using Mann-Whitney U test). (C) Control experiment in which the internalization step (Intenaliz.) was performed at 4 °C. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Please click here to view a larger version of this figure.
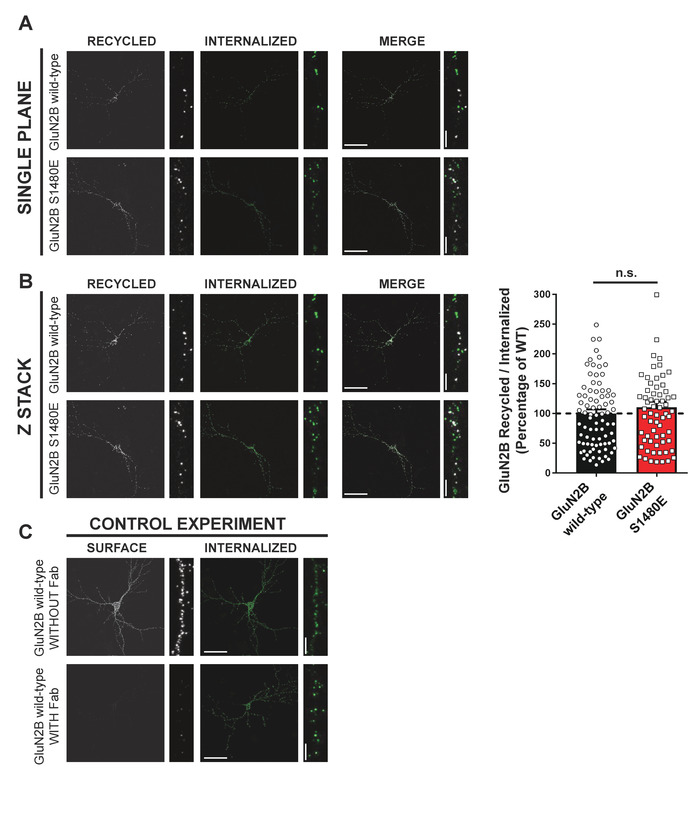
Figure 4: Phosphorylation of GluN2B at S1480 does not modify NMDAR recycling. Primary hippocampal neurons were transfected with either GFP-GluN2B WT or the phospho-mimetic mutant GFP-GluN2B S1480E on DIV11-12 as shown in Figure 3. Following 3-4 days of protein expression, surface GFP was labeled on live cells with a rabbit anti-GFP antibody and cells were then returned to 37 °C to allow for receptor internalization by endocytosis. Remaining surface-expressed receptors were blocked by Fab incubation and recycling was allowed for 45 min. Available surface expressed exogenous receptors (recycled) were visualized with Alexa 555-conjugated secondary antibody, and the internalized population identified after permeabilization using Alexa 647-conjugated antibody. For clarity, surface GFP-GluN2B is pseudocolored in white and internalized GFP-GluN2B is pseudocolored in green. (A) Single plane and (B) Z-stacked (maximum intensity projection) confocal pictures. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Graph shows the lack of effect the GluN2B S1480 phosphorylation has on recycling. Recycling index: recycled receptors/internalized receptors (n = 5; number of cells: WT = 27; S1480E = 24; values represent mean ± SEM; n.s. = non-significant using Mann-Whitney U test). (C) Control experiment in which the Fab incubation step to block surface-expressed epitopes was skipped. Scale bars = 50 µm (whole cell) or 5 µm (dendrite). Please click here to view a larger version of this figure.
