Here, we have used the tubule squash method to visualize transitory cell populations undergoing the prophase to metaphase I (G2/MI) transition, which were enriched by harvesting testes from juvenile wild-type mice undergoing the first wave of spermatogenesis (24 dpp). Figure 1 depicts representative images of the various cell stages that can be visualized using the tubule squash method. Enriched populations of metaphase I spermatocytes were visualized using antibodies against alpha-tubulin (Figure 1A). We utilized markers for early and late meiosis I to stage meiotic progression. Tubule squash preparations were immunolabeled with an antibody to the synaptonemal complex protein, SYCP3, to visualize stages of prophase I. Leptotene/zygotene and pachytene/diplotene spermatocytes are shown in Figure 1B. Spermatocytes undergoing the first meiotic division can be clearly identified using antibodies against alpha-tubulin (Figure 1A and 1C). To study late meiotic events, tubule squashes were immunolabeled using antibodies against the cell cycle kinase Aurora B (AURKB), which localizes to the inner centromere during metaphase, then relocalizes to the spindle midzone and cleavage furrow during anaphase and cytokinesis, respectively (Figure 1C). Cytological features of chromosomes were visualized during the first meiotic division. To observe ICDs, tubule squash preparations were immunolabeled using antibodies against the meiosis-specific alpha-kleisin subunit of cohesin, REC8 (Figure 1D). Chromosome dynamics during anaphase were assessed using alpha-tubulin immunolabelling and the DNA stain, DAPI. A spermatocyte successfully completing the first meiotic division is shown in Figure 1E, while a spermatocyte containing an anaphase bridge is shown in Figure 1F. Using the tubule squash technique, meiotic spindle length, chromosome alignment and segregation can be measured and compared between mutant and control mice.
The utility of this method for visualization of additional cell-cycle transitions and subcellular structures associated with prophase I is demonstrated in Figure 2. During the leptotene substage of prophase I, initiation of homologous chromosome pairing is mediated by dynamic changes in telomere attachments to induce formation of chromosome clusters facing a single pole of the nuclear envelope (NE)16,17,18,19,20. This conformation, known as the chromosome "bouquet" is thought to increase the likelihood and frequency of inter-homolog interactions (Figure 2A). Initiation and completion of synapsis between homologous chromosomes occurs during the zygotene (Figure 2B) and pachytene substages (Figure 2C), respectively, corresponding with progressive dispersal of the chromosome bouquet. To simultaneously assess synapsis and chromosome bouquet dynamics in juvenile mice, we immunolabeled tubule squash preparations with antibodies to SYCP3 and the centromeres, and stained for DNA using DAPI. Mouse chromosomes are telocentric, therefore centromere immunolabeling was used to detect changes in telomere NE attachments.
Meiotic sex chromosome inactivation (MSCI) is a hallmark unique to male meiosis, whereby the non-homologous X and Y chromosomes circumvent checkpoint activation by undergoing chromosome-wide phosphorylation of histone variant H2AFX (yH2AX; pSer139)21,22,23. The X-Y chromosome pair is transcriptionally silenced and compartmentalized into a dense nuclear subdomain called the "sex body." Spermatocytes undergoing MSCI can be readily identified during the pachytene and diplotene substages by immunolabeling tubule squash preparations with antibodies to yH2AX (Ser139). Figure 2D depicts a spermatocyte at the pachytene substage immunolabelled with antibodies against yH2AX (Ser139), the centromeres and stained with DAPI to visualize the DNA.
Centrosome duplication is semi-conservative and occurs during prophase I of male meiosis upon completion of DNA repair to ensure that each resulting daughter cell inherits two centrioles (one centrosome) following cell division24,25. Centrosomes consist of two orthogonally arranged centrioles connected by a flexible linker and are surrounded by an amorphous matrix of proteins known as the pericentriolar material/matrix (PCM)26. After duplication in the pachytene substage, newly formed centriole pairs disengage from one another and undergo centrosome maturation and migration to opposite poles during the diplotene stage to facilitate formation of bipolar spindles. An early diplotene spermatocyte undergoing disjunction of newly formed centriole pairs was visualized by immunolabeling tubule squash preparations with antibodies against pericentrin (PCNT), a protein component of the PCM and Centrin-3 (CETN3) to mark centrioles (Figure 2E). Staging was determined using antibodies against SYCP3, and DAPI was used to stain DNA.
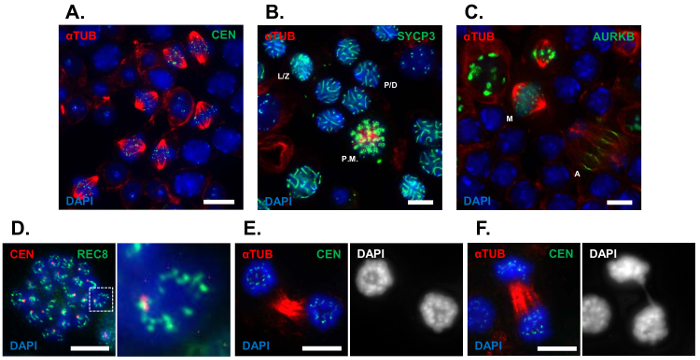
Figure 1: Representative analysis of the meiotic G2/MI transition using tubule squash preparations. Tubule squash preparations were performed on seminiferous tubule segments of C57BL/6J mice aged 24 dpp. (A) Representative field of developing spermatocytes immunolabeled with antibodies against alpha-tubulin (red), centromeres (green) and the DNA stain DAPI (4', 6-diamidino-2-phenylindole, blue). (B) Wild-type tubule squashes were stained with DAPI (blue) as well as immunolabeled with antibodies against alpha-tubulin (red), and the lateral element of the SC, SYCP3 (green). (C) Wild-type tubule squashes were stained with DAPI (blue) as well as immunolabeled with antibodies against alpha-tubulin (red), and the cell cycle kinase AURKB (green). Cytological features of chromosomes such as ICDs and anaphase bridges can be visualized using tubule squash preparations. (D) To observe ICDs, prometaphase spermatocytes were immunolabeled with antibodies against centromeres (red) as well as a meiotic specific cohesin component, REC8 (green) and stained with DAPI (blue). (E, F) Chromosome morphology was visualized during anaphase using antibodies against alpha-tubulin (red), centromeres (green), and the DAPI stain (blue). A full complement of developing spermatocytes can be identified using the tubule squash technique (L/Z, leptotene/zygotene stage spermatocytes; P/D, pachytene/diplotene stage spermatocytes; P.M., prometaphase spermatocytes; M, metaphase spermatocytes; A, anaphase spermatocytes). Scale bar = 10 µm. Please click here to view a larger version of this figure.
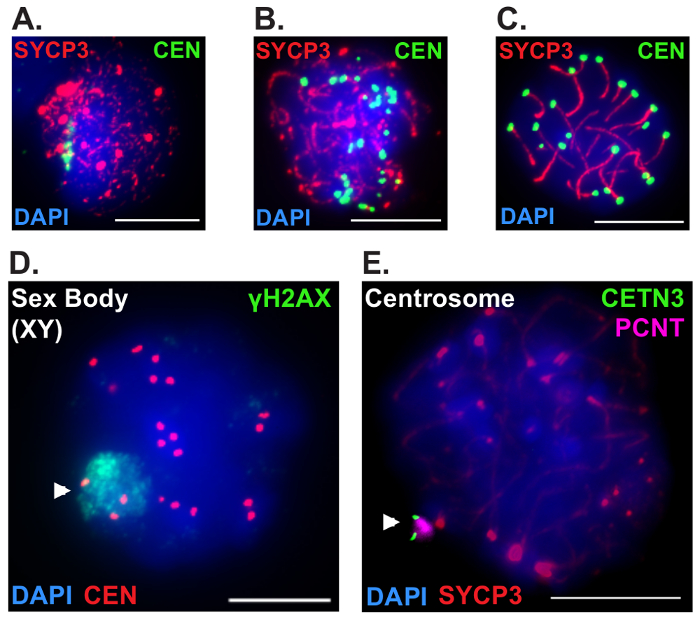
Figure 2: Examples of additional morphological changes and structures associated with meiotic prophase progression in males using tubule squash preparations. Tubule squash preparations were performed on seminiferous tubule segments of C57BL/6J mice aged 12-18 dpp and were stained with DAPI (4', 6-diamidino-2-phenylindole, blue) to label DNA. (A-C) Homolog synapsis is coupled to the formation and progressive dispersal of the chromosome "bouquet" during prophase progression. Leptotene (A), zygotene (B), and pachytene (C) stage spermatocytes were immunolabeled with antibodies against SYCP3 (red) and the centromeres (green). (D-E) Tubule squash preparations can be used to detect unique cytological changes and structures during male prophase I progression including meiotic sex chromosome inactivation (MSCI) and centrosome duplication. (D) Pachytene stage spermatocytes undergoing MSCI were detected using antibodies to yH2AX (Ser139) to label the sex-body (green, arrowhead), and centromeres are shown in red. (E) Diplotene stage spermatocytes were identified using antibodies against SYCP3 (red), and centrosomes (arrowhead) were detected using antibodies against Centrin-3 (green) and Pericentrin (magenta) to label the centrioles and the pericentriolar material/matrix, respectively. Scale bar = 10 µm. Please click here to view a larger version of this figure.
| Item | Amount | Final Concentration |
| 1x PBS | 10 mL | 1x |
| 16% PFA | 500 μL | 0.8% (v/v) |
| 10% TritonX-100 in PBS | 100 μL | 0.1% (v/v) |
Table 1: Fix/lysis solution. Description: Adjust pH to 9 with NaOH [50mM]. Wrap the container of solution in aluminum foil to protect from light and store at 4 °C. Do not use the fix/lysis solution if more than a week old. Solid PFA can also be used to make the solution. Use of a fumehood is recommended to avoid PFA exposure.
| Item | Amount | Final Concentration |
| 1x PBS | 50 mL | 1x |
| BSA | 1.5 g | 3% (w/v) |
| Horse Serum | 5 mL | 10% (v/v) |
| 10% TritonX-100 in PBS | 250 μL | 0.05% (v/v) |
Table 2: Antibody dilution buffer (ADB) recipe. Description: Store ADB at 4 °C or freeze stocks at -20 °C if making larger quantities. ADB can become contaminated, make sure good aseptic techniques are used and assess the solution for contamination prior to each use. Prepare smaller aliquots of ADB to minimize contamination.
| Cell type | Cellular Process | Mouse Age (dpp) |
| Leptotene | DNA DSB formation | 10 – 12 |
| Assembly of axial elements | ||
| Zygotene | Initiation of homologous DSB repair and synapsis | 12 – 16 |
| Pachytene | Completion of autosomal DSB repair | 16 – 20 |
| Maturation of crossover recombination events | ||
| Complete synapsis between homologs | ||
| Meiotic Sex Chromosome Inactivation (MSCI) | ||
| Centriole Duplication/Centrosome Maturation | ||
| Diplotene and Diakinesis | SC Desynapsis | 18 – 22 |
| Centrosome separation | ||
| Metaphase I | Spindle Checkpoint | 22 – 26 |
| Round Spermatid | Protamine replacement | >24 |
| Acrosome formation |
Table 3: Near-synchronous first wave of spermatogenesis in juvenile mice. Description: Specific stages of spermatogenesis are enriched at different stages of juvenile mouse development.
