Protrusion formation during T cell migration is a highly dynamic process, which is actin dependent. To visualize protrusion formation of primary human CTL, we transiently transfected a mEGFP fused protein to label the actin cytoskeleton in CTL as described before11. One day after transfection, the cells were embedded in the collagen matrix. Image stacks were acquired every 40 s with a step-size of 1 µm at 37 °C using light-sheet microscopy. As shown in Figure 2A and Supplementary Movie 1, during migration, human CTL form lemon-shaped major protrusions, fringed by fine spindle-like structures. In the experiment shown here, not only one single cell was imaged but a large volume (450 x 450 x 538 µm3) (Figure 2B). Thus, the optical quality and resolution shown here were obtained with a low magnification objective (20X / 1.0 DIC VIS IR). Therefore, the resolution could be further improved with higher NA objectives.
In the experiment shown in Figure 2 and Movie 1, CTLs were automatically tracked as described in the protocol Part 4. The trajectories of CTL are depicted in Figure 2B. Two parameters of migration were analyzed: velocity and persistence. Persistence is defined as the displacement divided by the total track length. The analysis shows that the mobility of CTL is very diverse in 3D: the velocities range from 0.01-0.19 µm/s with an almost 20-fold difference, and the persistence ranges from 0 – 0.7 (Figure 2C). It was reported in mice that in vivo interstitial migration average velocity of T cells was 4 µm/min16. Recently, we have determined the average velocity of CTL to be 4 – 5 µm/min11 using the methods described in this paper. These results demonstrate that light-sheet microscopy is a powerful tool to visualize cell behavior, for example, cell migration and cell-cell interaction. In vivo, many factors and a non-homogeneous ECM of the complex composition including soluble factors that can affect migration will modify T cell behavior.
Application of a collagen matrix is not limited to live cells. Fixation and immunostaining can also be performed in the matrix. Figure 3 shows an example of fixed samples in 3D collagen gel, in which endogenous perforin1 (PFN1) and actin were stained. The sample was illuminated either from a single side (Single side illumination) or from both sides (Dual side illumination). We observe that cells at different z-positions are evenly stained, indicating that procedures described in our protocol achieve satisfactory penetration of antibodies into collagen gels. More importantly, after fixation CTL exhibited the same morphology as in live cell imaging (compare Figure 3 and Figure 2B), indicating that also the morphology is well maintained by this protocol. In addition, illumination from the single side or from both sides does not make a significant difference in the quality of images at the focal plane. Considering that less area is exposed to the laser with the mode of single side illumination compared to dual side illumination, single side illumination is more recommended.
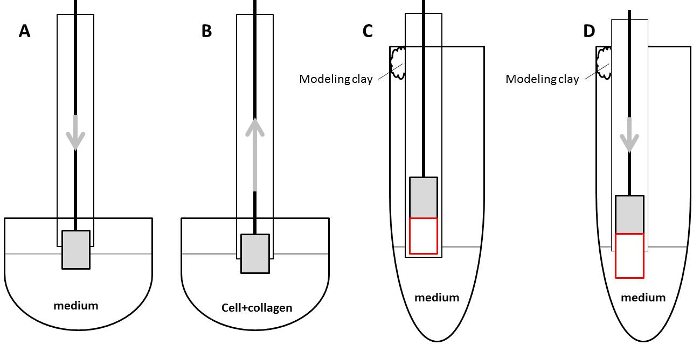
Figure 1: Illustration of handling capillaries during sample preparation. (A) Put the plunger into the capillary and how to wet the plunger with the culture medium. (B) Pull the cell/collagen mix into the capillary. (C) Mount capillaries using modeling clay. The plunger and the collagen rod are depicted in gray and red boxes, respectively. (D) Handle collagen rod for equilibration with the culture medium or for immunostaining. The plunger and the collagen rod are depicted in gray and red boxes, respectively. Please click here to view a larger version of this figure.
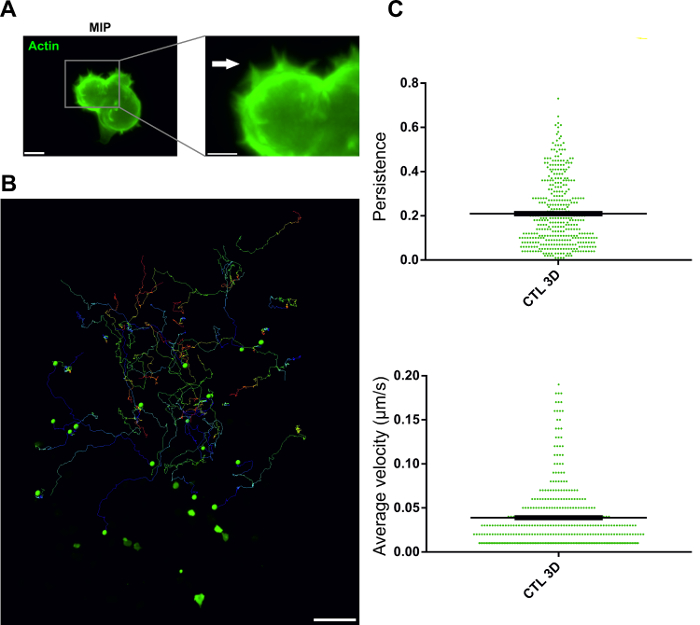
Figure 2: Tracking primary human CTL in 3D collagen and visualizing actin dynamics. (A) Maximum intensity projection of actin in primary human CTL. Actin was labeled in CTL as previously described11. Day one post transfection cells were embedded in 0.5 % collagen matrix. One representative cell is shown. Scale bars are 5 µm. Fine actin structures at the protrusion edge are highlighted by the arrowhead. (B) Trajectories of CTL for 6 h. The trajectories are automatedly tracked by the software (Color code: blue = start, red = end of the track, 450 × 450 × 538 µm3 volume). The scale bar is 50 µm. (C) Quantification of velocity and persistence. One dot represents one cell. Results are shown as mean ± SEM from 4 donors. Please click here to view a larger version of this figure.
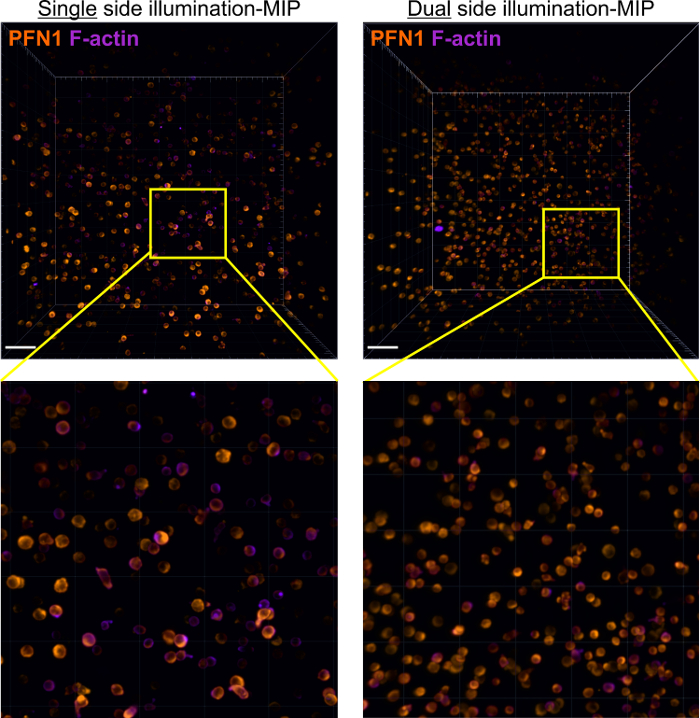
Figure 3: Immunostaining of endogenous profilin1 (PFN1) and actin in CD8+ T cells embedded in the 3D collagen matrix. Primary human CD8+ T cells were embedded in 0.25% collagen. After PFA fixation the sample was stained using anti-PFN1 (orange) and phalloidin (purple) and visualized by light-sheet microscopy at a step-size of 1 µm for a total volume of 440 x 440 x 1,000 µm3. The sample was illuminated either from a single side (Single side illumination) or from both sides (Dual side illumination). Maximum intensity projections (MIP) from 3D reconstruction are shown. The scale bars are 50 µm. Please click here to view a larger version of this figure.
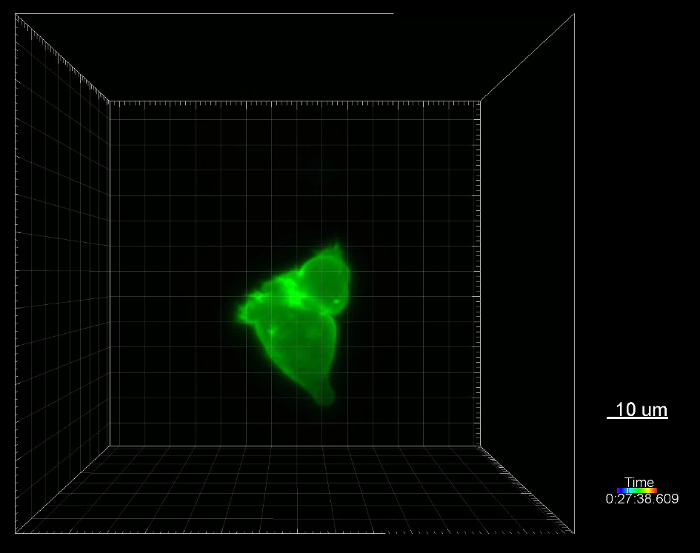
Movie 1: Generation of protrusions during T cell migration. The cell shown in the movie was taken from the volume shown in Figure 2A. Cells were embedded in a collagen matrix (0.5%). Images were acquired every 40 s for 6 h at 37 °C using light-sheet microscopy. Please click here to view this video. (Right-click to download.)
