1. Preparing stock solutions and reagents
- Prepare dithiothreitol (DTT) stock solution by dissolving crystalline DTT in distilled water to a final concentration of 1000 mM. Adjust the pH to 7.0 with 1 M NaOH solution. Aliquot and store at -20 °C.
- Prepare ATP stock solution by dissolving crystalline ATP in distilled water to a final concentration of 100 mM. Adjust the pH to 7.0 with 1 M NaOH solution. Aliquot and store at -20 °C.
- Prepare 10x NADH buffer containing 70 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 10 mM MgCl2, 0.9 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 3 mM NaN3. Adjust the pH to 7.0 with 1 M NaOH solution. Store at 4 °C.
- Prepare 1x myosin buffer containing 10 mM MOPS and 0.1 mM EGTA. Adjust the pH to 7.0 with 1 M NaOH solution. Store at 4 °C. Add bovine serum albumin (BSA) and DTT to a final concentration of 0.1% (w/v%) and 1 mM, respectively, before use.
- Prepare 1x actin buffer containing 4 mM MOPS, 0.1 mM EGTA, 2 mM MgCl2, and 3 mM NaN3. Adjust the pH to 7.0 with 1 M NaOH solution. Store at 4 °C. Add BSA and DTT to a final concentration of 0.1% (w/v%) and 1 mM, respectively, before use.
- Prepare NADH stock solution by dissolving crystalline NADH in 10x NADH buffer to a final concentration of 5.5 mM. Aliquot and store at -20 °C.
- Prepare PEP stock solution by dissolving crystalline PEP in 10x NADH buffer to a final concentration of 50 mM. Aliquot and store at -20 °C.
- Prepare LDH stock solution by dissolving lyophilized LDH powder in a mixture of glycerol and 10x NADH buffer (50%:50%) to a final concentration of 2000 U/mL. Centrifuge the solution to remove any undissolved protein present (7,197 x g, 20 °C, 10 min). Transfer the supernatant into a clean centrifuge tube carefully. Aliquot and store at -20 °C.
- Prepare PK stock solution by dissolving lyophilized PK powder in a mixture of glycerol and 10x NADH buffer (50%:50%) to a final concentration of 10000 U/mL. Centrifuge the solution to remove any undissolved protein present (7,197 x g, 20 °C, 10 min). Transfer the supernatant into a clean centrifuge tube carefully. Aliquot and store at -20 °C.
- Reconstitute the lyophilized cardiac and skeletal muscle myosin II samples by adding 100 µL distilled water to obtain 10 mg/mL stock solutions corresponding to ~37.9 µM and ~40.8 µM myosin concentrations (monomeric), respectively. For further details, see manufacturer's instructions.
- Prepare F-actin from rabbit muscle acetone powder as described by Pardee and Spudich25.
2. Measuring ATPase activities and inhibitory effects of small molecule inhibitors
- Prepare compound plate.
- Dissolve compounds of interest in high-quality dimethylsulfoxide (DMSO).
- Create fifteen-step serial 1:2 dilutions starting from 10 mM compound concentration in DMSO.
- Transfer the samples to a 384-well polypropylene plate in triplicates (12.5 µL each) using a multichannel pipette. Use two rows on the compound plate for one compound (instead of three columns) to minimize the number of wells potentially affected by edge effects. Use the last three wells in the second row for each compound as negative control (DMSO only). Do not use the first and the last row on the plate for compound dilutions.
- Transfer pure DMSO into the wells of the first row (reserved for NADH calibration).
- Use the last row for positive control.
NOTE: Para-aminoblebbistatin at 4 mM concentration in DMSO was used here.
- Prepare 4500 µL of 20 µM diluted actin solution for each assay plate (384-well black-wall polystyrene microplate) by diluting actin stock solution in actin buffer. Mix the solution thoroughly by pipetting up and down 30x using a 5 mL pipette to reduce viscosity and heterogeneity by breaking actin filaments. Centrifuge the solution to remove any precipitated protein present (7,197 x g, 20 °C, 10 min). Carefully transfer the supernatant into a clean centrifuge tube.
- Prepare master mix containing LDH and PK enzymes ("enzyme mix"). For each assay plate, combine 171.4 µL of LDH solution, 171.4 µL of PK solution and 3189.3 µL or 3252.9 µL of myosin buffer for assays involving cardiac or skeletal muscle myosin II's, respectively, in a 15 mL conical centrifuge tube. Do not add any myosin at this point to avoid aggregation and precipitation.
- Prepare master mix containing all substrates ("substrate mix"). For each plate, combine 162.1 µL of ATP, 162.1 µL of PEP and 324.1 µL of NADH solution in a 15 mL conical centrifuge tube. Do not add actin at this point to avoid aggregation and precipitation.
- Create seven-step serial 1:2 dilutions of NADH for calibration starting from 250 µM.
- Mix 12.3 µL of NADH stock solution with 257.7 µL of myosin buffer in a 1.5 mL microcentrifuge tube.
- Aliquot 135 µL of myosin buffer into seven 1.5 mL microcentrifuge tubes.
- Transfer 135 µL of solution from the first tube into the second and mix by pipetting. Repeat until reaching the 7th tube.
- Use the last tube as no-NADH control (buffer only).
- Using an 8-channel pipette, transfer 20 µL of the NADH calibration solutions into the first row of the assay plate in triplicates.
- Add 68 µL of cardiac or 4.2 µL of skeletal muscle myosin II to the enzyme mix. Vortex briefly.
- Except the first row, dispense 8.4 µL of the prepared myosin-enzyme mix into each well of the assay plate using an automated dispenser.
- Transfer 100 nL of solutions from the compound plate to the assay plate containing enzyme mix using an automated liquid handling system equipped with a 100 nL pin tool head.
- Shake the assay plate for 1 min at room temperature at 1200 rpm using a microplate shaker.
- Add 4,052 µL of the centrifuged actin solution to the substrate mix. Vortex briefly.
- Dispense 11.6 µL of actin-substrate mix into each well of the assay plate (except first row) to start the enzymatic reaction using an automated dispenser.
- Shake the assay plate for 1 min at room temperature at 1200 rpm using a microplate shaker.
- Centrifuge the assay plate at 101 x g for 30 s.
- Make sure that the inner temperature of the plate reader has been stabilized at 25 ˚C. Load the plate and shake for another 30 s. This shaking step is necessary to make the shape of the liquid surface similar in each well and allows time for the plate to reach measurement temperature.
- Record NADH fluorescence for 30 min scanning the plate in 45 s intervals. Use a 380 nm, 10 nm bandwidth excitation filter and a 470 nm, 24 nm bandwidth emission filter in conjunction with a 425 nm cut-off dichroic mirror. Run the measurement in high-concentration mode. Optimize the number of flashes, detector gain, plate dimensions and measurement height before running the assays.
NOTE: Final assay conditions are 300 nM cardiac/20 nM skeletal muscle myosin II, 10 µM actin, 40 U/mL LDH, 200 U/mL PK, 220 µM NADH, 1 mM PEP, 1 mM ATP in a buffer containing 10 mM MOPS (pH = 7.0), 2 mM MgCl2, 0.15 mM EGTA, 0.1 mg/mL BSA, 0.5% (v/v) DMSO and 1 mM DTT. The total volume is 20 µL/well. The highest final compound concentration is 50 µM. 20 µM para-aminoblebbistatin in 0.5% DMSO serves as the positive control and 0.5% DMSO alone is the negative control. All measurements are carried out in triplicates.
3. Analyzing data
- Plot the observed fluorescence intensity against time for each well.
- Perform simple linear regression to determine the slope and intercept of the fluorescence responses for each well. The slope is proportional to the ATP (NADH) consumption rate, while the intercept is proportional to the NADH concentration at the beginning of the measurement (t = 0 s).
- Construct a calibration curve for NADH by plotting the intercepts obtained for the first row of the plate against the concentration of NADH. Make sure that the intercepts depend linearly on the NADH concentration.
NOTE: The intercepts estimate the real fluorescence intensities at t = 0 s with much more confidence than the average of the raw fluorescence intensity reads at t ≈ 0 s. - Perform simple linear regression to obtain the slope and intercept of the NADH calibration line.
NOTE: The intercept describes the fluorescence background signal (no NADH present), while the slope corresponds to the extrapolated/theoretical fluorescence intensity of a 1 M NADH solution in that particular experiment. - Divide the slope of the fluorescence response obtained for the rest of the wells by the slope of the NADH calibration line to convert fluorescence changes to ATP consumption rates.
- Plot the ATP consumption rates against the concentration of the inhibitor.
- To determine inhibitory constants, use appropriate statistical software to fit the dose-response data to the following quadratic equation corresponding to a simple one-to-one binding equilibrium model:

where Y is the ATP consumption rate, Ymin is the ATP consumption rate int the absence of inhibitor, Ymax is the theoretical ATP consumption rate at 100% inhibition, KI is the inhibitory constant, [E]t and [I]t are the total concentration of the enzyme (myosin) and inhibitor, respectively.
The typical plate layout map used for screening experiments is shown in Figure 1. The first and last rows are reserved for NADH calibration and positive control (20 µM para-aminoblebbistatin, 0.5% DMSO), respectively. The remaining rows (B to O) are used to test the inhibitory activity of compounds. Here, fifteen-step serial 1:2 dilutions starting from 10 mM compound concentration in DMSO are prepared and transferred from the compound plate to the assay plate, such that the highest final compound concentration is 50 µM (in 0.5% DMSO) on the assay plate. Two rows are used to obtain a dose-response curve for one compound (48 datapoints/compound). Note that the plate layout maps can be re-designed to support the specific aims of a given project. For example, if the goal were to obtain single-point screening data for a large number of compounds, one could test 112 compounds on a single 384-well plate using the same layout for positive control and NADH calibration (calculating with triplicates for each compound). It is always advised to have a minimum of 3 datapoints for one compound (or for each concentration) and to avoid using only the wells along the edges of the plate for one compound, as these datapoints may be influenced by edge effects. To estimate the importance of edge effects, always run a full plate with negative control only first.
The fluorescence intensities have a linear dependence on the concentration of NADH as shown in Figure 2A. The slope of the linear fit is used during data analysis to convert fluorescence changes to reaction rates. Note that the raw fluorescence intensity trace obtained for each well of the NADH calibration is analyzed by linear regression first (a similar analysis is shown in Figure 2B,C for compound data). These traces are expected to show exponential decay over time due to photobleaching of the fluorophore. However, photobleaching is very slow and therefore, the raw data can be analyzed by linear fits. The slope and intercept of these fits correspond to the initial rate of photobleaching and the fluorescence intensity at t = 0 s, respectively. The intercepts of these linear fits are used instead of the average of the raw fluorescence reads at t = 0 s to construct the NADH calibration curve because the intercepts are estimated based on more data and, therefore, the associated errors are much smaller.
Figure 2B,C demonstrates that regardless of the myosin used or the presence of the inhibitor, the time courses are linear in the time window of the measurements. The highest (50 µM) and lowest (0 µM) inhibitor concentrations here correspond to ~100% and 0% inhibition, respectively. Note that due to the amount of raw data, the actual analysis would appear chaotic if shown on a single panel. Therefore, these panels have been simplified to better visualize the process. The average of the raw fluorescence intensity reads was calculated for all of the parallel experiments (triplicates for each concentration) and converted to NADH concentrations here. Only 3 inhibitor concentrations are shown. In the real analysis, each raw fluorescence intensity trace (48/compound tested) is analyzed by linear regression first, and subsequently, the slopes are converted to ATP consumption rates.
It is always advisable to demonstrate that the reaction rates change linearly with the enzyme concentration, as shown in Figure 2D and Figure2E for skeletal and cardiac muscle myosin II's, respectively. Based on the linear fits, the final assay concentration of the enzyme can be easily estimated. For example, a reaction rate of ~5 x 10-8 Ms-1 is recommended for 30-min time courses. If an activator is used in the reaction mixtures (such as actin here), it is recommended to run the experiments both in the presence and absence of the activator to ensure that the expected effect (activation) is present. The conditions and the procedure must follow the final protocol as closely as possible. Here, a dilution series of myosin was prepared in myosin buffer in eight microcentrifuge tubes first. Subsequently, a mix of LDH and PK enzymes were added. Finally, the reactions were started by adding substrate mix to each tube in parallel, using a multichannel pipette. Reaction mixes were immediately transferred to one row of the assay plate in triplicates. If actin was absent, actin buffer was used instead. No other parameters were changed (see the note of step 2.16 in the protocol for final assay conditions).
Figure 2F shows ATP consumption rates obtained for multiple negative and positive control reactions (half plate each). These data can be compared based on the Z` value or "screening window coefficient"26, which is a widely used statistical parameter to estimate the quality of high-throughput assays. It compares the positive and negative controls by taking both the means and the standard deviations into account:
 ,
,
where σn, σp and µn, µp are the standard deviations and means of the negative and positive controls, respectively. The two populations are well separated if the Z` value falls between 0.5 and 1. A Z` = 0.78 obtained here shows that the assay can be considered as excellent26.
To demonstrate that the assay can be used to determine inhibitory constants, the small molecule myosin inhibitor blebbistatin8 and two analogues, para-nitroblebbistatin12 and para-aminoblebbistatin13 have been chosen here, as shown in Figure 3A and Figure 3B. Blebbistatin is an uncompetitive, allosteric myosin inhibitor27,28. One molecule of blebbistatin binds to one motor domain of myosin and blocks the ATPase cycle by stabilizing the myosin-ADP-phosphate complex27,28. Therefore, the inhibitory effects of blebbistatin derivatives was modelled using a simple, one-to-one binding model here (see step 3.7 in the protocol). Note that this model may not have been applicable if the ATP consumption rates had not shown linear dependency on myosin concentration (see Figure 2D,E). The kinetic aqueous solubility of blebbistatin, para-nitroblebbistatin and para-aminoblebbistatin has been reported to be 426 µM, 3.6 µM and 9.3 µM, respectively13. No abnormalities in the signal were observed at or below the reported solubilities in our experiments; however, several artifacts appeared when either blebbistatin or para-nitroblebbistatin was used above their reported solubility values, as shown in Figure 4. Therefore, the signal recorded at concentrations higher than the solubility was excluded from the data analysis in these cases. Para-aminoblebbistatin is highly soluble, and therefore, solubility was not a limiting factor in that case.
Finally, it is always recommended to test whether the inhibitory effects of any positive hits are specific to the target ATPase enzyme. The coupled reaction system employs two other enzymes, LDH and PK, and inhibition of one of these would result in a false positive signal. Running the ATPase assay with an unrelated ATPase enzyme may help to filter out these false positive hits (for further recommendations, see discussion). Para-aminoblebbistatin and apyrase, an ATP hydrolyzing enzyme producing ADP and inorganic phosphate29, were used here as an example to demonstrate such a control experiment, as shown in Figure 5.

Figure 1: Assay plate layout. Seven-step serial 1:2 dilutions of NADH starting from 250 µM concentration is prepared and subsequently dispensed into row A in triplicate for calibration (black to green color gradient). The last three wells of row A contain myosin buffer only (no NADH control, white). The last row (P) is used for the positive control (20 µM para-aminoblebbistatin; red). A typical dose-response experiment requires two rows (e.g., B and C). Therefore, 7 dose-response experiments can be run in parallel on a single 384-well plate (represented by blue to white color gradients). Every sample is loaded as triplicates. Here, the highest final compound concentrations start at 50 µM (in 0.5% DMSO). The last three wells of every second row are reserved for the negative control (no compound, 0.5% DMSO only; cyan). Please click here to view a larger version of this figure.

Figure 2: Representative ATPase data. (A) A two-fold dilution series of NADH was prepared and transferred into the first row of each measurement plate. Fluorescence intensity was recorded for 30 min and the raw data was analyzed by simple linear regression. The intercept of each regression line was plotted against the concentration of NADH. Note that in an ideal case, the fluorescence intensity at t = 0 s could simply be used to obtain the calibration line. However, while the raw fluorescence data is very noisy, the intercepts give an accurate estimate of the fluorescence intensity at t = 0 s and their associated standard error (shown as error bars) is very small. (B,C) Representative fluorescence intensity traces of the skeletal (B) and cardiac (C) muscle myosin II ATPase reactions were recorded in the presence of various levels (see insets) of para-aminoblebbistatin. For simplicity, data points and error bars represent the average of three independent measurements and the associated standard deviation, respectively. Simple linear regression was performed (solid lines) to obtain reaction rates. Note that a typical dose-response experiment is comprised of 15 different inhibitor concentrations and negative control in triplicates on the measurement plate (see Figure 1) and the linear regression is performed individually for each fluorescence intensity trace. For simplicity, only 3 different concentrations are shown here. (D,E) Basal (red) and actin-activated (blue) ATPase rates were determined for various skeletal (D) and cardiac (E) muscle myosin II concentrations. The ATPase rates show linear dependency on the myosin concentration. (F) Positive (red) and negative (blue) controls (half plate each) were run in parallel on a 384-well assay plate and the Z-factor (Z`) was calculated to assess the quality of the ATPase assay. A Z' value of 0.78 indicates a reliable assay with very well separated positive and negative controls. Please click here to view a larger version of this figure.

Figure 3: Dose-response curves and analysis of inhibitory constants. Cardiac (A) and skeletal (B) muscle myosin II's were used to test the inhibitory activity of blebbistatin, para-aminoblebbistatin and para-nitroblebbistatin. ATPase rates (blue) were obtained by applying simple linear regression to the raw fluorescence data. Error bars represent the standard error of fitting and were used as weighting factors during fitting a quadratic equation (see step 3.7 in the protocol) representing a simple equilibrium binding model (red). Data obtained above solubility was influenced by artifacts and excluded from analysis. Please click here to view a larger version of this figure.
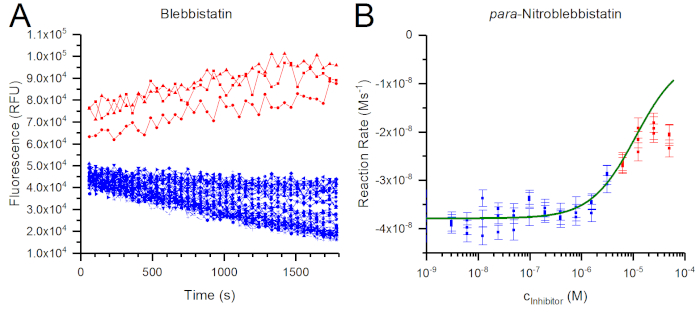
Figure 4: Solubility-related artifacts. (A) Fluorescence intensity traces for blebbistatin obtained in an ATPase assay using skeletal muscle myosin II show linearly decreasing signal depending on the amount of inhibitor present (blue). However, when blebbistatin was used above solubility (50 µM initial blebbistatin concentration), an increase in the signal was observed (red), most likely due to the formation of brightly fluorescent blebbistatin crystals13. (B) In the case of para-nitroblebbistatin, which is a non-fluorescent analog of blebbistatin12, the raw fluorescence intensity traces appeared normal (decreasing). However, the highest level of inhibition was much lower than expected (based on the positive control). Therefore, only the reaction rates obtained below solubility (blue) were included in data analysis. Reaction rates obtained above solubility (red) diverge from the determined dose-response curve (green), as precipitation limits the amount (concentration) of the inhibitor remaining in solution. Please click here to view a larger version of this figure.

Figure 5: Inhibitory effects of para-aminoblebbistatin in skeletal muscle myosin II and apyrase ATPase assays. Para-aminoblebbistatin inhibited skeletal muscle myosin II with a KI of 1.7 µM, while no inhibition was detected when apyrase29 was used as an ATPase in the same coupled reaction system. This experiment clearly demonstrates that para-aminoblebbistatin is specific to myosin and does not inhibit PK or LDH, thus the detected inhibitory effect is not an artifact. Apyrase was used at 0.5 nM concentration. No myosin or actin was present, and the reaction was performed in a buffer containing 100 mM MOPS (pH = 7.0), 3 mM CaCl2, 2 mM MgCl2, 3 mM NaN3, 1 mM DTT, and 0.1% BSA. No other modifications were made to the protocol. Please click here to view a larger version of this figure.
