All methods described here have been published elsewhere and contain small modifications from the original methods.
1. Wastewater collection and sample pre-processing
NOTE: Due to the low concentrations of SARS-CoV-2 RNA in environmental samples, the implementation of a concentration step is crucial for a successful detection33,34,35. Described here is the first reported method for the detection of SARS-CoV-2 in wastewater36.
- Collection and concentration of wastewater sample(s)
- Collect 1 L of 24 h composite wastewater sample. Store the sample at 4 °C and proceed with the protocol within 24 h.
- Homogenize the sample by gently shaking the bottle. Collect 70 mL into a 100 mL centrifugal tube or divide the sample into two centrifugal tubes of 50 mL.
- Spike 20 µL of mengovirus (3.2 x 103 copies/µL) to 70 mL of each sample as an internal control of the concentration process. Alternatively, spike 10 µL of mengovirus (3.2 x 103 copies/µL) into each 50 mL centrifuge tube.
- Homogenize the sample and centrifuge at 700 x g for 10 min to remove large particles and organisms (pellet).
- Use the resulting supernatant for concentration with centrifugal ultrafiltration devices with a cutoff of 10 KDa by centrifuging at 4,000 x g for 40 min at 4 °C. Elute the concentrate by centrifuging at 700 x g for 40 min and inverting the position of the ultrafiltration device.
- Measure the volume of the resulting concentrate. The volume will change depending on the quantity of solids in the sample that clog the centrifugal filters. In this study, the concentrate volumes obtained were between 200-1,200 µL.
- Use the resulting concentrate for RNA extraction using a commercial kit or an in-house method. For the samples used in this study, a specific commercial kit for viral RNA extraction with an elution volume of 50 µL is used.
- Measure the concentration of the extracted RNA with a fluorometric RNA quantification kit. Divide the extracted RNA into aliquots and freeze at -80 °C until further analysis.
NOTE: For each RNA isolation procedure set, a negative control of isolation (NCI) should be included, containing only buffers to detect possible contamination during the extraction.
- Collection of air samples using a Coriolis compact air sampler
- Choose a sampling strategy according to the research goal by defining the sampling frequency and sampling locations.
- Set the flow rate on the air sampler (here, 50 L/min for 30 min). Please note that a flow rate of more than 200 L/min can degrade viral RNA, so it is recommended to use a flow rate below 200 L/min.
- Place a sterile cone in the air sampler before starting the collection by pulsing the start. Once the sampling is finished, add 5-15 mL of sterile phosphate-buffered saline (PBS) into the cone. Gently vortex or shake the sample by hand for 15 s. Store the samples for up to 24 h at 4 °C or at -80 °C in cryotubes.
- An optional concentration step can be performed. Clean and decontaminate the cones after each experiment using bleach.
- Extract RNA using a commercial kit or an in-house method. For the samples in this study, a specific commercial kit for viral RNA extraction with an elution volume of 50 µL is used.
- Measure the concentration of the extracted RNA with a fluorometric RNA quantification kit. Divide the extracted RNA into aliquots and freeze at -80 °C until further analysis.
NOTE: For each RNA isolation procedure set, a negative control of isolation (NCI) should be included, containing only buffers to detect possible contamination during the extraction.
2. Quantification of SARS-CoV-2 RNA by real-time-quantitative polymerase chain reaction (RT-qPCR)
NOTE: The below protocol is according to the CDC 2019-Novel Coronavirus (2019-nCoV) RT-PCR diagnostic panel37. Divide the primer/probe mix into several aliquots to avoid freezing and thawing cycles.
- Prepare the reaction mastermix for each target (mengovirus; SARS-CoV-2 [N1 and N2 genes]) as in Table 1, in a clean hood in the reagent setup room. Mix the primers/probe mix and enzyme by inversion five times. Alternatively, light pulse-vortex the primers/probe mix and enzymefive times.
NOTE: Keep all reagents cold during preparation and use. In order to minimize freeze-thaw cycles, it is highly recommended to freeze in aliquots. - Spin down (1,000 x g for 15-30 s) the tubes to collect the contents at the bottom and place the tubes in a cold rack or on ice.
- Label 1.5 mL microcentrifuge tubes for each target. Add to each microcentrifuge tube the amount of each reagent needed (volume per reaction times the number of reactions including the needed controls). Mix by pipetting up and down and centrifuge for 5 s to collect the contents at the bottom.
- Keep the microcentrifuge tubes in a cold rack and dispense 15 µL into strip PCR tubes or a 96-well plate in a cooling rack. Cover the plate and move it to the nucleic acid handling area (keep it in a cooling rack).
- Thaw an aliquot of extracted RNA and gently vortex for 5 s. Pipette 5 µL of RNA (in addition to 5 µL of non-template control [NTC], negative control of isolation [NCI], and positive control [PC]) to each reaction well or tube containing the previously prepared mastermix. Change gloves often as needed to avoid cross-contamination.
- Cover the entire reaction plate or tubes and gently vortex. Centrifuge (1,000 x g for 15-30 s) the plate or PCR tubes.
- Start the 25 µL RT-qPCR with the following cycling conditions: reverse transcriptase at 45 °C for 10 min; polymerase activation at 95 °C for 10 min; 45 cycles of denaturation at 95 °C for 15 s; and annealing/extension at 60 °C for 45s.
- Calculate the recovery efficiency of mengovirus controls as reported by Conte et al.38:
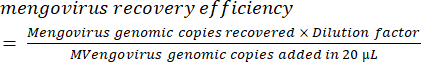 × 100
× 100
3. Sequencing variants in wastewater and data analysis
NOTE: The described protocol is a modified protocol created by Quick et al.39,40. It uses two sets of primers for SARS-CoV-2 genome amplification by PCR tiling methodology-ARTIC primers and VarSkip primers. A combination of primers is used to guarantee the best genome coverage and to minimize the possibility of novel mutations causing primers of one type to fail. In general, the protocol is divided into three parts: reverse transcription (RT) and amplicon generation, sequencing library preparation, and sequencing and data analysis.
- Reverse transcription and amplicon generation
- Ensure the input samples have a known qPCR cycling threshold (Ct) value to correctly dilute them in PCR-grade water. The Ct value is obtained during the quantification with qPCR. The dilutions are as follows: if the Ct value is 12-15, dilute the samples 1:100; if the Ct value is 15-18, dilute the samples 1:10; if the Ct value is 18-35, do not dilute the sample. Samples above a Ct value of 35 have a high chance of not working.
- In a PCR plate, pipette 16 µL of each sample to its position on the plate. Add 4 µL of reverse transcription (RT) mastermix (5x). Cover the plate and start the PCR reaction with the following conditions: 25 °C for 2 min, 55 °C for 20 min, and 95°C for 1 min.
- Prepare 10 µM dilutions of each primer set (ARTIC pool A and pool B, and VarSkip pool A and pool B). Prepare the mastermix for each set of primers (ARTIC set A and B, and VarSkip set A and B) as in Table 2. Four mastermixes will result from this.
- On a new PCR plate, pipette 20 µL of the mastermix to each corresponding well. Add 5 µL of the RT sample to each mix.
NOTE: Each sample will have four reaction mixtures-ARTIC pool A and B, and VarSkip pool A and B. - Cover the plate and start the PCR reaction as follows: polymerase activation at 98 °C for 30 s; 35 cycles of denaturation at 98 °C for 15 s; and annealing/extension at 65 °C for 4 min. Spin down (1,000 x g for 15-30 s) the plate. Combine the ARTIC reaction pools and separately combine the VarSkip reaction pools. Each sample will have two 50 µL amplicon reactions.
- Add 50 µL of bead-based reagent for PCR purification to each well and mix well by pipetting. Incubate at room temperature for 5 min.
NOTE: Before every use, mix the PCR purification reagent vigorously to resuspend the beads. - Place the plate on a magnetic separation stand and wait for the beads to form a pellet and the liquid to clear (~5 min). Pipette and discard the supernatant. Keep the PCR plate on the magnetic stand.
- Maintaining the PCR plate on the magnetic stand, add 200 µL of 80% ethanol to each sample without touching the bead pellet. Remove the ethanol.
- Repeat the previous step. Leave the plate on the magnetic stand uncovered for 30 s to allow the ethanol to evaporate.
- Remove the plate from the magnetic stand and add 15 µL of PCR-grade water to each sample. Resuspend the pellet by pipetting. Incubate at room temperature for 2 min.
- Place the plate back on the magnetic stand and allow the beads to pellet (2 min). Carefully pipette 15 µL of the supernatant from each sample to a new PCR plate.
- Measure the concentration of each sample with a fluorometric RNA quantification kit . Take approximately 50 ng of each sample in 12.5 µL to the next step. If required, dilute the sample in PCR-grade water.
- Library preparation
- Add 1.75 µL of reaction bufferfrom the Illumina DNA library preparation kit and 0.75 µL of enzyme mix to each sample. Cover the plate, vortex briefly, and spin down (1,000 x g for 15-30 s). Incubate at 21°C for 5 min and at 65 °C for 5 min.
- In a new plate, pipette 3 µL of PCR-grade water for each sample to a new PCR plate. Add 0.75 µL of the prepared samples, 1.25 µL of barcodes for sequencing library preparation, and 5 µL of T4 DNA ligase mastermix. Mix well by pipetting, briefly spin down (1,000 x g for 15-30 s) in a centrifuge and incubate at 21 °C for 20 min and at 65 °C for 10 min.
NOTE: If using less than 25 samples, double all the volumes in this step. - Pool all the samples together by adding 10 µL of each sample to the same low-binding tube. Take 480 µL to the next step.
- Add 192 µL of bead-based reagent for PCR purification to the pool and mix well by pipetting. Incubate at room temperature for 10 min.
- Place the tube on a magnetic stand and wait for the supernatant to clear and a bead pellet to form. Remove the supernatant. Remove the tube from the magnetic stand.
- Add 700 µL of the short fragment buffer (SFB) and mix by pipetting. Place back on the magnetic stand and wait for the pellet to form and the liquid to clear (~5 min). Pipette out the supernatant and discard it.
- Repeat the previous step. Leave the tube on the magnetic stand. Add 100 µL of 80% ethanol without touching the bead. Pipette out the ethanol and allow the bead to dry for 30 s.
NOTE: Do not allow the bead to over-dry. - Remove the tube from the magnetic stand and add 35 µL of PCR-grade water. Mix by pipetting and incubate at room temperature for 2 min. Place the tube on the magnetic stand and allow the bead to pellet and the liquid to clear. Pipette 35 µL of the supernatant to a new tube.
- Measure the RNA concentration of the pooled library. Pipette the volume needed to reach an amount of 30-50 ng of RNA and fill it with PCR-grade water to reach a final volume of 30 µL.
- Prepare the adapter ligation reaction mix: 30 µL of the pooled library, 5 µL of Adapter Mix II, 10 µL of the ligation reaction buffer (5x), and 5 µL of T4 DNA ligase. Mix by pipetting and spin-down (1,000 x g for 15-30 s). Incubate at room temperature for 10 min.
- Add 20 µL of the bead-based reagent for PCR purification and mix by pipetting. Incubate for 10 min at room temperature.
- Place the tube on the magnetic stand and wait for the bead pellet to form and the supernatant to become colorless. Carefully pipette out and discard the supernatant.
- Remove from the magnetic stand and add 125 µL of SFB. Mix by pipetting and place back on the magnetic stand to separate the beads. When the liquid is clear, pipette and discard the supernatant.
- Repeat the previous step. Leave the tube open on the magnetic stand for 30 s for some of the leftover liquid to evaporate.
- Remove the tube from the magnetic stand and add 15 µL of the elution buffer. Mix well by pipetting and briefly spin-down (1,000 x g for 15-30 s). Incubate at room temperature for 5 min. Place on the magnetic stand for 2 min.
- Pipette 15 µL of the supernatant into a new low-binding tube. This is the final library. Measure the concentration with a fluorometric DNA quantification kit.
NOTE: In the next step, 12 µL is needed. If the concentration is very high and less than 12 µL of the library is needed, fill up to 12 µL with the elution buffer reagent.
- Flow cell loading and sequencing
- Insert the flow cell (R9.4.1) into the real-time DNA and RNA sequencing device.
- Prepare the priming mix by adding 30 µL of flush tether (FLT) reagent to a tube of flush buffer (FB) reagent. Vortex to mix and spin down (1,000 x g for 15-30 s).
- Open the priming port cover. Using a 1,000 µL pipette, set to 200 µL and insert the tip into the priming port. Turn the volume setting wheel and increase the volume until the liquid in the tip is seen. Discard the tip.
NOTE: Only turn the wheel until a few µL of liquid in the tip is seen. Pulling larger amounts could damage the flow cell. - Slowly add 800 µL of the priming mix to the priming port, taking care not to introduce bubbles. Wait for 5 min.
- In the meantime, prepare the libraries for loading. Mix 37.5 µL of the sequencing buffer, 25.5 µL of the loading buffer, and 12 µL of the library.
NOTE: Mix the loading buffer before pipetting. The beads in this reagent settle very quickly. - Open the port cover. Pipette 200 µL of the priming mix into the priming port.
NOTE: While pipetting into the priming port with the cover port open, one will notice small drops coming from the sample port. Pipette slow enough so the sample port can pull the drops in without spilling over. - Before loading the prepared library, mix well by pipetting. Using a 100 µL pipette set to 75 µL, take the library and pipette drop by drop into the sample port. Do not touch the port and wait for each drop to be absorbed into the port before loading the next drop to avoid overflowing of the liquid.
- Close the cover port and priming port. Open the software and start the sequencing experiment. Select basecalling On. For a library with 96 multiplexed samples, 10-12 h of sequencing is usually enough to generate sufficient data.
NOTE: Using the software, one can insert the sample sheet in .csv format. The sample sheet requires the following data: flow_cell_id, position_id, sample_id, experiment_id, flow_cell_product_code, and kit. The flow cell ID is needed (listed on the side of the flow cell) together with the kit used. For the suggested protocol, the LSK109 kit should be selected.
- Sequencing data analysis
- Insert the compressed fastq reads into the wf-artic workflow41. Run the workflow separately with two different primer schemes (ARTIC/V4.1 and NEB-VarSkip/v1a). Two ". primertrimmed.rg.sorted.bam" files are created for each sample.
- Merge the BAM files into a single file with the "samtools merge" command42. Insert the merged BAM file into Freyja tool, leaving default settings, to recover relative lineage abundances43.
- To create a FASTA file, insert the merged BAM file into the "samtools mpileup" and "ivar" tools with default settings44,45. For mutation calling, load the FASTA file into the Nextclade tool46,47.
The results summarized in Table 3 show examples of the detection and quantification of SARS-CoV-2 RNA in wastewater and air samples using the method described in this article. Wastewater samples were collected from wastewater treatment plants in Spain and Slovenia and were considered positive if the Ct was less than 40 in at least two of the three replicates, with quantification considered valid if the Ct had a variation of less than 5%. In Spain and Portugal, indoor and outdoor air samples were collected, and the same rules were applied. Duplicates were used for the air samples, not triplicates as for the wastewater samples, as the aim of the study was to detect SARS-CoV-2 RNA and not to quantify it. Furthermore, an RT-qPCR run was only considered valid if all controls used (as described in this protocol) behaved as expected and if no significant deviations were observed in the mengovirus RNA control. For these samples, the mengovirus recovery efficiency varied between 13.7% and 19.7%. The inhibition was assessed by diluting tenfold and 100-fold extracted RNA and by comparing the resulting RT-qPCR results, as well as using a commercial kit. The instructions for each inhibition control kit should be followed as described by the manufacturers.
Wastewater samples had a standard deviation from 1.91% to 13.98% among the triplicates and ranged from 3.05 x 103 to 2.83 x 108 gene copies/L. Air samples ranged from 6.17 x 103 to 5.48 x 109 with a standard deviation between the duplicates between 0.54% and 10.95%. This is in accordance with previous studies6,48,49,50.
As described in this protocol, the samples were sequenced and an example of three sequenced sample results is summarized in Table 4 and Figure 1. In all three samples, the lineage BA.5.x was assigned as the most prevalent by the Freyja tool (95.3%, 95.4%, and 99.8%).
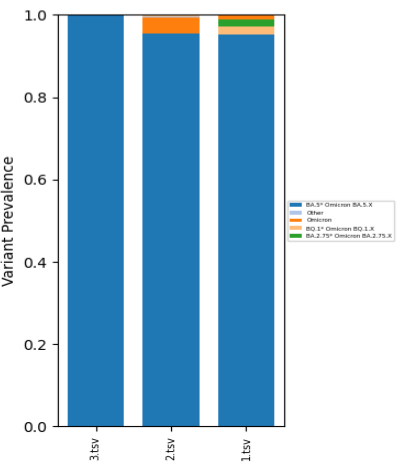
Figure 1: SARS-CoV-2 lineage prevalence in selected wastewater samples. Lineages were assigned using the Freyja tool. The summary of prevalence numbers does not necessarily reach 100%, as some of the data is not of sufficient quality. Please click here to view a larger version of this figure.
Table 1: Reagents and volumes to be added of each to prepare the mastermix for SARS-CoV2 quantification by RT-qPCR. Please click here to download this Table.
Table 2: Reagents and volumes to be added of each to prepare the mastermix to amplify the complete SARS-CoV-2 genomes in the samples. Please click here to download this Table.
Table 3: Summary of RT-qPCR quantification results of SARS-CoV-2 RNA from wastewater and air samples. The N1 gene was used for quantification and the N2 was used for confirmation (positive or negative). The results are expressed as copies/L. Please click here to download this Table.
Table 4: Detected mutations and lineage prevalence. Lineages were assigned using the Freyja tool and according to the mutations of interest found using Nextclade. Genome coverage for each sample is shown. The summary of prevalence numbers does not necessarily reach 100%, as some of the data is not of sufficient quality. Please click here to download this Table.
