Table 1: Appropriate volumes of papain, albumin-ovomucoid, and final suspension used in dissociation steps. The appropriate volumes of papain and albumin-ovomucoid to be used with various numbers of ventral midbrain and ventral spinal cord tissues were modified from the manufacturer's instructions after several rounds of optimization. Because tissues are subject to stress during dissociation and sorting, a pooled collection of more than 10 ventral midbrains and more than three ventral spinal cords is recommended. The volume of papain was determined by considering the balance between effective dissociation and the stress of this procedure. The volume of albumin-ovomucoid inhibitor solution is half of that of papain. The appropriate volume of Hibernate E final suspension was determined such that cell density does not exceed 107 cells/mL, but the cells do not become excessively diluted.
| Number of midbrains (X) | Papain | Albumin-ovomucoid | Hibernate E |
| 10 ≤ X ≤20 | 200 μL | 100 μL | 600 μL |
| 20 < X ≤30 | 300 μL | 150 μL | 700 μL |
| 30 < X ≤40 | 400 μL | 200 μL | 800 μL |
| Number of spinal cords (Y) | Papain | Albumin-ovomucoid | Hibernate E |
| 3 ≤ Y ≤5 | 200 μL | 100 μL | 500 μL |
| 5 < Y ≤10 | 400 μL | 200 μL | 800 μL |
| 10 < Y ≤15 | 600 μL | 300 μL | 1200 μL |
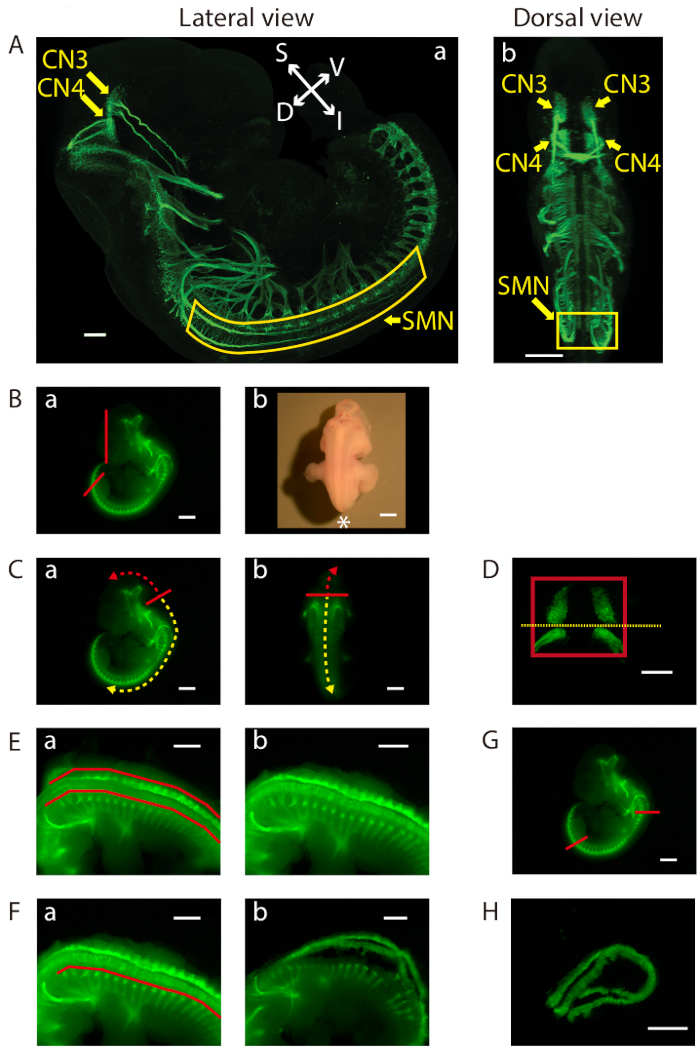
Figure 1: Dissection of the ventral midbrain and the cervical (C1)-lumbar (L2-L3) portion of the ventral spinal cord. (A) Lateral (a) and dorsal (b) views of GFP-positive motor neurons in an E11.5 IslMN:GFP transgenic mouse embryo under fluorescein isothiocyanate (FITC) illumination. A whole mount E11.5 embryo was prepared in order to make the embryo transparent. Subsequently, the embryo was analyzed by immunofluorescence labeling with anti-GFP staining (green). Images were captured under a confocal microscope. Scale bars = 200 µm (lateral view) and 400 µm (dorsal view). Abbreviations: S = superior; I = inferior; V = ventral; D = dorsal. (B-H) Dissection steps highlighted on images of E11.5 ventral midbrain and ventral spinal cord tissues taken with an equipped camera under bright light (Bb) or FITC illumination using a fluorescence dissection stereomicroscope. Scale bars = 200 µm (D), and 1 mm (A−C, E−H). (B) (a) Removal of the face and tail of the embryo by cutting along the red lines. (b) Embryo positioned for dissection. Positioning of the front of the microscope is indicated by an asterisk. (C) Cutting along the solid red line in order to slit open the roof of the fourth ventricle (a) lateral view and (b) dorsal view. Use of this opening to cut along the surface of the embryo dorsal to the brain (trajectory indicated by dashed red arrow). This exposes the tissue containing mesenchyme, CN3, and CN4, which can be lifted out of the cranium. For SMN dissection, insertion of forceps into the same opening between the fourth ventricle and its roof, then cutting toward the caudal side of the embryo (trajectory indicated by dashed yellow arrow). (D) Final view of the ventral midbrain containing bilateral GFP-positive CN3 and CN4 nuclei. The edges of the tissue are highlighted by a red rectangle. Cutting along yellow dotted line to collect CN3 and CN4 nuclei separately, if desired. (E) After opening the rest of the hindbrain and spinal cord, flapping dorsal tissues pinched off above the red lines on both sides with tweezers (a) before, and (b) after. (F) Bilaterally removal of excess tissue ventral to the spinal cord along the red line (a) before, and (b) after. (G) Cutting of the ventral spinal cord at the two locations indicated by the red lines. On the rostral side, cutting of the floating ventral spinal cord transversely above C1 where the first GFP-positive anterior horn projects. Cutting of the caudal end of the spinal cord transversely at the upper boundary of the lower limb. Once these cuts are made, the cervical (C1) through lumbar (L2-L3) portion of the ventral spinal cord can be dissected away. (H) Final view of the ventral spinal cord containing GFP-positive SMN columns.
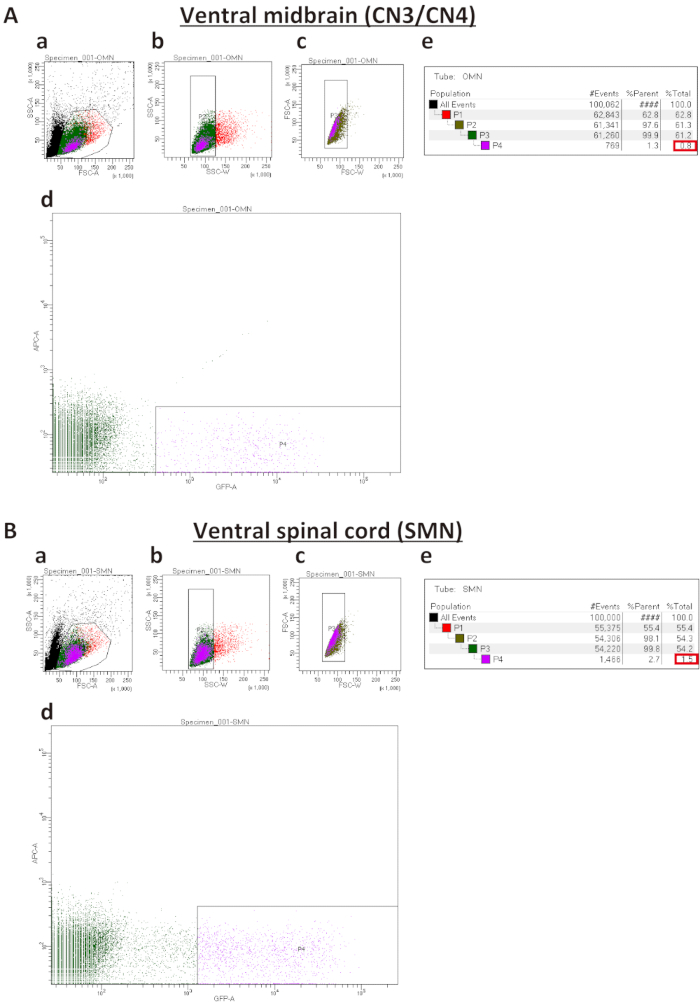
FIGURE 2. Representative sort plots of ventral midbrains (A) and ventral spinal cords (B). (Aa and Ba) Forward Scatter Area (FSC-A) versus Side Scatter Area (SSC-A) sorted plot before exclusion of debris and dead cells. (Ab, Bb, Ac, Bc) Sorted plots for exclusion of cell clumps (b) and doublets (c) based on Width (SSC-W) versus SSC-A and Forward Scatter Width (FSC-W) versus FSC-A, respectively. (Ad and Bd) Sorted plots to isolate IslMN:GFP -positive motor neurons. In order to obtain a pure culture, the GFP gate must be set higher for SMNs (Bd) than for CN3s/CN4s (Ad). (Ae and Be) Percentages of cells gated for collection by FACS sorting. %Parent represents the percentage of cells in the current gated population relative to the number of cells in the previous gated cell population, whereas %Total represents the percentage of gated cells relative to total cells. Expected percentages of GFP-positive cells as compared to total cells (boxed in red) are 0.5−1.5% for CN3/CN4 and 1.5−2.5% for SMN. If the dissection was performed successfully, these percentages can be used as a benchmark to set up the GFP-positive gate in (Ad and Bd).
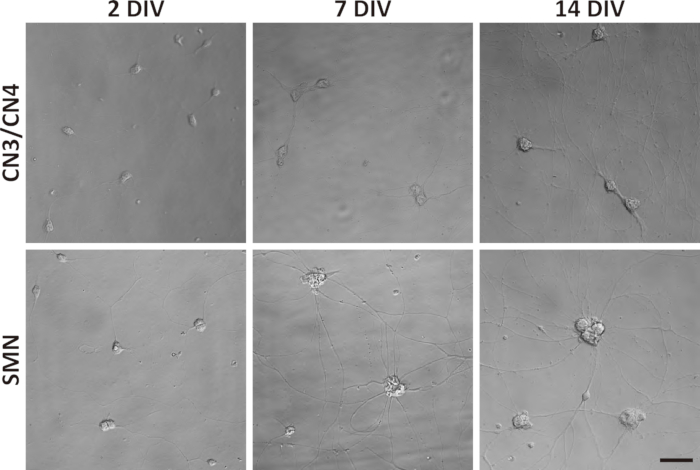
Figure 3: Phase-contrast images of primary CN3/CN4 and SMN monocultures at 2, 7, and 14 DIV. Representative differential interference contrast images of primary CN3/CN4 and SMN cultures were captured at 2, 7, and 14 DIV with inverted fluorescence microscope using corresponding image acquisition and processing software and 40x objectives. Neuronal processes became thicker and longer by 14 DIV. Neuronal cell body sizes became enlarged and tended to aggregate in long-term cultures, especially for SMNs. Both cultures can be maintained at least 14 DIV. Scale bar = 50 µm.
