Figure 1 summarizes the procedure for tissue isolation, preparation and culture on MEA. We show the consecutive steps of tissue dissection to isolate the spiral ganglion (SG) from the sensory epithelium of the organ of Corti (OC) and stria vascularis (SV) and annexed spiral ligament (Figure 1 A–C). Spiral ganglion explants (3-4 in number) are cut with micro-scissors from the ganglion as schematically illustrated in Figure 1D and placed on MEA (Figure 1E), over the electrode occupied area (2.2 mm2). The OC is placed in proximity, outside of the electrode surface. The growth of the culture can be monitored over time (Figure 1G). A schematic diagram of the protocol is shown in Figure 1F. Electrophysiological activity can be detected after 6 days of culture, which increase with prolonged culture time. We recommend assessing 18 day cultures which produce significantly higher numbers of recording electrodes compared to earlier time points 5.
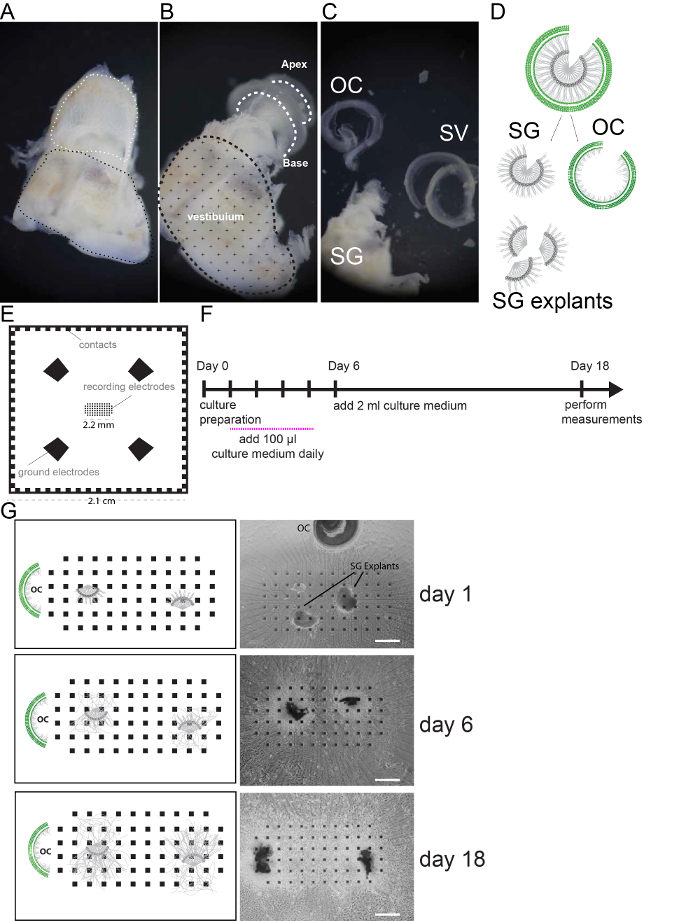
Figure 1. Cell culture preparation. (A) Freshly dissected mouse inner ear. White dashed line indicates the location of the cochlea, black dashed line indicates the vestibular region. (B) Mouse inner ear after removal of the cochlear bony wall. Cochlear turns are indicated by white dashed lines. (C) The Spiral ganglion (SG) and modiolus, the organ of Corti (OC) and the stria vascularis (SV) and spiral Ligament are shown after dissection. (D) Schematic of SG and OC dissection and SG explant preparation {figure adapted from 5 with approval from the publisher}. (E) Illustration of the multi electrode array used in this study. Recoding electrodes are organized in a rectangular grid in the center and occupy an area of 2.2 mm2. 4 Ground electrodes and side contacts are illustrated. (F) Schematic of the culture protocol. Recordings are performed at day 18. (G) Representative pictures (bright field images) and schemes of SG explants on MEA monitored in culture at day 1, 6 and 18. Scale bars = 400 µm. Please click here to view a larger version of this figure.
Spontaneous activity can be detected on MEA and shown as a raster plot where each line of the plot represents a detected spike. A representative example showing spontaneous activity detected from several electrodes (different colors in the graphs) is shown (Figure 2A, 2B, and 2C).
Additionally, MEA electrodes can be used to stimulate the culture. A representative example showing a biphasic pulse used for stimulation (Figure 2D), 5 responding electrodes (red traces) and non-responding electrodes (blue traces) is shown in Figure 2E. For these experiments one electrode was used for stimulation and all the other electrodes for recording. Single action potentials appeared 1 msec after the stimulation artifact (black arrow head). The MEA can be immunostained at the end of the procedure in order to assess the coverage of the neuronal processes over the electrode area. The electrode indicated in green in Figure 2F was used for stimulation, and the electrodes indicated in red were used to record responses (Figure 2E).
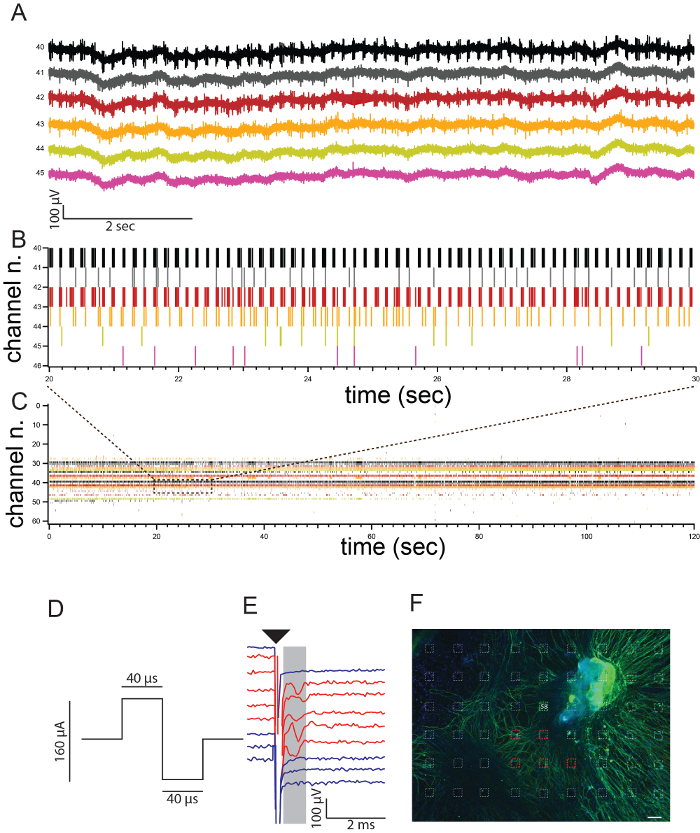
Figure 2. Data recordings on MEA. (A) Traces of original recordings of six out of 63 electrodes showing spontaneous activity. (B) Raster plot of the six electrodes of Figure 2A after spike detection. Each bar represents one action potential. (C) Raster plot including all electrodes (as in A and B) Activity is recorded from 63 electrodes (channels number 0-63) for 2 min. (D) A biphasic stimulus with total duration of 80 µsec and amplitude of 80 µA was used for stimulation of the culture from one electrode (E58 in Figure 2F). (E) Representative example of raw data traces obtained after stimulation from electrode 58 showing action potentials (red traces) or without responses (blue traces) after stimulation (black arrow-head). (F) Spiral ganglion culture on MEA immunostained for the neuronal marker TUJ (green) at the end of the experiment to visualize neuronal coverage of the electrode area {figure adapted from 5 with approval from the publisher}. Electrode 58 used for stimulation is indicated in green, responding electrodes are indicated in red. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Finally, the MEA setup allows for studying possible electrode surface modification that may increase recording sensitivity. Two commercially available MEA electrodes were used in this study with two different platinum surfaces: Grey Platinum (Grey Pt), consisting of a 150 nm thick Pt layer (impedance: 400 KΩ/1 KHz) and Black Platinum, (Black, Pt), obtained by electrochemical deposition of Pt at the end of the micro-fabrication process (impedance: 20 KΩ/1 KHz). See Materials Table (point 6) for details.
Six independent MEA experiments were performed per electrode type. Black Pt MEA allows for detection of neuronal activity on a higher number of electrodes per MEA (Figure 3A) and reduction of the stimulus amplitude needed to elicit a response (Figure 3B). We analyzed for 30-35 independent electrode pairs, in 6 different experiments, the current threshold needed to achieve a response. Black Pt electrodes performed significantly better showing a threshold of 31.09 µA +/-2.4 compared to Grey Pt electrodes 47.57 µA +/- 1.97.
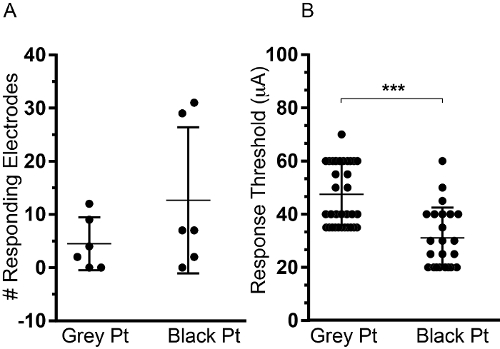
Figure 3. Comparison of Grey vs Black Pt electrodes. Two types of electrodes (Grey and Black Pt) were compared side by side using 12 independent MEA cultures, 6 on each MEA type. (A) The total number of responding electrodes per experiment is shown. (B) Amplitude threshold to elicit a response, measured using 30-35 independent electrode pairs in 6 independent MEA experiments is shown. Data are shown as Mean+/-SD. (Student t Test p <0.001) Please click here to view a larger version of this figure.
