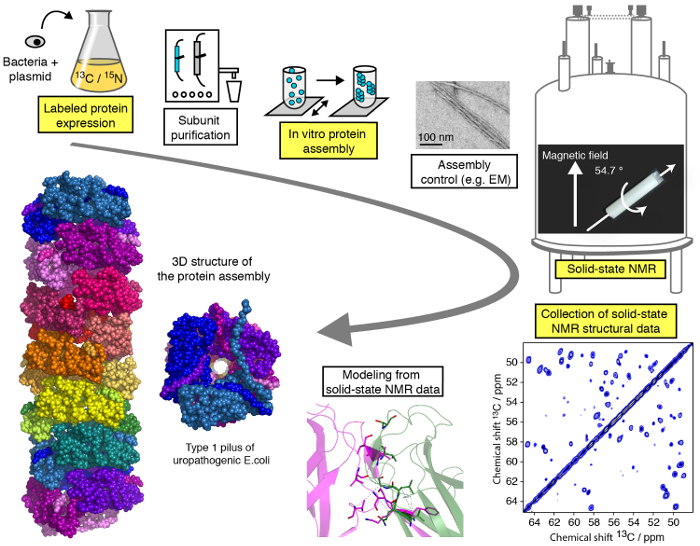
1. Solid-State NMR échantillon productiona Production d’uniformément 13 C / 15 sous-unités de protéines marquées N contenant du plasmide utilisation fraîchement transformée, peptide-pET24-6his E. coli BL21 (DE3) cellules, récolter de la gélose préparés. Ensemencer une pré-culture 15 mL de milieu LB préchauffé pour une colonie. Incuber à 37 ° C, avec 200 tr/mn agitation du jour au lendemain. Transférer le pré-culture dans 1 L de la culture principale de préchauffé M9 moyen (voir composition dans le tableau 1), qui contient les sources requis de carbone et d’azote isotopes marqués. Remarque : Ceci peut être uniformément 13 C-étiqueté glucose, sélectivement (1 – 13 C)- ou (2 – 13 C)-étiqueté glucose 29 , 30 , 31 ou () 1, 3 – 13 C)- ou (2 – 13 C)-étiqueté glycérol 32 , 33. Ceci incuber à 37 ° C et mesurer l’ OD 600 dès que la culture devienne trouble. Lorsque l’ OD 600 a atteint une valeur de 0,8, induire d’expression de la protéine avec 0,75 mM IPTG pour 4 h. Remarque : Les conditions optimales d’induction peuvent varier d’une protéine à l’autre. Récupérer les cellules par centrifugation pendant 30 min à 6000 x g et 4 ° C. Croquis de purification (non montré dans la vidéo protocole) lyser les cellules à l’aide de techniques standard telles que le traitement de sonication ou lysozyme et de purifier la sous-unité de la protéine à l’aide de techniques établies ou adaptés à la étudié l’Assemblée protéine. Remarque : La protéine peut être exprimée en corps d’inclusion, vérifiez pour la localisation des protéines par lyse cellulaire et centrifugation ultérieure. Si la protéine se trouve dans les sédiments avec les débris cellulaires, elle a été accumulée dans le corps d’inclusion. Test expression et la pureté de l’échantillon de protéine par exemple par une standard 12-15 % Tris-Tricine SDS-PAGE, voir Figure 2 a. Si la pureté est suffisante, les protéines peuvent être assemblés, sinon effectuer une étape de purification supplémentaire. In vitro l’assemblage des filaments protéiques Vérifier la concentration de protéines dans la solution purifiée. Si la protéine contient des résidus absorbants, mesurer l’absorption dans un spectrophotomètre UV à 280 nm. Insérez le tampon pur dans le spectrophotomètre comme étalonnage et mesurer puis l’absorption de protéine. Calculer la concentration de la protéine à l’aide du coefficient d’absorption de la sous-unité de la protéine avec la Loi de Beer-Lambert adaptée : A λ = ε λ x l densité x c ; ici, A est l’optique à une longueur d’onde donnée λ ; ε est la coefficient d’absorption spécifique ; l est la longueur de la trajectoire optique en cm ; c est la concentration en mol/L. Concentré de la protéine à une concentration de 1 mM dans une unité de filtration centrifuge. Introduire l’échantillon dans l’unité de filtration centrifuge et centrifuger à 4000 x g pendant 30 min, mélanger la solution dans l’unité de filtre doucement avec une pipette pour éviter les dépôts de protéines au niveau du filtre. Répétez la procédure jusqu’à ce que la concentration désirée soit atteinte. Remarque : La concentration optimale de montage dépend du système et doit être optimisée (généralement entre 0,1 – 1 mM). Si un équimolaire mixte étiqueté échantillon de deux lots de différentes protéines marquées est prêt (par exemple (50/50) (U – 15 N)-/ (U – 13 C)-étiqueté), mélange doit avoir lieu avant ou juste après l’étape de concentration afin d’assurer homogénéisation de la solution mélangée. Transférer l’échantillon dans un tube et incuber sous agitation pendant une semaine à température ambiante il. Habituellement la polymérisation des sous-unités en filaments est accompagnée de la solution devient trouble. Vérifier la morphologie microscopique de fibrilles et l’homogénéité par exemple par microscopie électronique (6300 X – 45000 X grossissement), voir Figure 2 b. Centrifuger l’échantillon pendant 1 h à 20000 x g et 4 ° C. Retirer la majorité du liquide surnageant, laissez seulement suffisamment de liquide pour couvrir la surface afin d’éviter le séchage des échantillons. Vérifiez le liquide surnageant des sous-unités non assemblé sur le spectrophotomètre UV. Laver l’échantillon en ajoutant H 2 O et prudent Resuspendre le contenu avec une pipette. Ne pas vortexer. Faire tourner l’échantillon pendant 30 min à 22000 x g et 4 ° C. répéter l’étape de lavage 3 fois. Vérifier pendant toutes les étapes de lavage si le culot conserve son aspect après centrifugation et le surnageant des sous-unités non assemblé sur le spectrophotomètre UV. Ajouter 0,02 % (p/v) d’azide de sodium (NaN 3) pour éviter la contamination bactérienne et de stocker l’échantillon jusqu’à la mesure à 4 ° C. Solid-State NMR rotor remplissage centrifuger deux fois pendant 30 min à 22000 x g et 4 ° C. Remove maximum de surnageant ; la consistance de l’échantillon qui en résulte dépend de l’assemblage des protéines et est habituellement une type gel dépôt. Utiliser une pipette capillaire pour introduire l’échantillon dans le rotor SSNMR. NOTE : Ici, nous présentons la procédure sur un rotor de diamètre 4 mm. Centrifugeuse centrifugeuse le rotor sur une table à introduire doucement l’échantillon dans le rotor. Répétez la procédure jusqu’à ce que le rotor est rempli. Maintenir un espace minimum pour fermer le rotor avec le capuchon du rotor. Si la quantité de l’échantillon n’est pas suffisante, introduire un encart disponible dans le commerce pour remplir l’espace restant pour assurer l’homogénéité de distribution d’échantillons dans le rotor. NOTE : Dépendant sur la consistance de l’échantillon assemblé, il serait plus convenable d’utiliser une spatule mince, surtout pour les échantillons très denses. Rotors de 2,5 à 4 mm de diamètre sont utilisés à des fréquences de MAS modérées. Ajouter des traces d’acide 4,4-dimethyl-4-silapentane-1-sulfonic (DSS) pour étalonnage interne de changement et de la température chimique. Fermez le bouchon avec un dispositif de fermeture. Vérifier sous une loupe si le bouchon est bien inséré et complètement fermé. figure 2 : représentant des résultats pour purification de protéine sous-unités et l’Assemblée. A) 15 % Tris-tricine SDS-PAGE de sous-unité protéique (y compris son 6-tag) à différentes étapes de purification. Piste 1 – marqueur de poids moléculaire de protéines ; piste 2 – e. coli BL21 (DE3) cellules contrôle non induit ; piste 3 – e. coli BL21 (DE3) cellules induites avec 0,75 mM IPTG ; piste 4 – solubilisée inclusions voie 5 – les fractions surnageantes des cellules lysat ; voie 6 – fraction purifiée après Nickel immobilisés par chromatographie d’affinité métallique (IMAC) FPLC et de dessalage. B) coloration négative des fibrilles protéiques par imagerie TEM. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure. 2. caractérisation structurale préliminaire basée sur unidimensionnelle (1D) à semi-conducteurs NMR mise en place initiale et 1D polarisation croisée (CP) Insérer le rotor en l’aimant de NMR commencent à Faites tourner le rotor à une fréquence de 5 kHz et attendre une stabilisation de ± 10 Hz, puis accélérer jusqu’à 7 kHz. Branchez-vous et correspondent à 1 H, 13 quater et 15 N canaux. Régler la température à 2-10 ° C (275-283 K) ; échantillon de chauffage à 7 kHz MAS est faible et le réglage de la température n’est habituellement pas nécessaire à cette fréquence MAS. Enregistrer un single-pulsé 1D 1 H du spectre en utilisant 16 balayages. Remarque : Des niveaux de puissance doivent ont précédemment été optimisées sur un composé de référence (comme un 13 C / < sup > 15 N histidine ou la glycine), afin de fournir des valeurs de départ permettant d’optimiser les niveaux de puissance dans les expériences sur l’échantillon cible ; Cette procédure est une tâche courante et donc hors du champ d’application du présent protocole. Garder la température au cours de toutes les expériences entre 2 et 10 ° C, dans des échantillons hydratés, que la température se reflète dans le déplacement relatif de la résonance de 1 H de l’eau en vrac en ce qui concerne le signal DSS 34. Mesurer la température suivant la relation δ (H 2 O) = 7.83-T/96,9 avec une précision de 1 à 2 ° C 35. Fréquence de MAS ensemble vers le haut la direction désirée et attendre jusqu’à stabilisation de ± 10 Hz. NOTE : Ici, il est démontré à une fréquence de 11 kHz. Tune et match 1 H et 13 C canaux à nouveau et ajuster la température à l’aide d’une expérience de 1 H 1 D. Configurer un 1D 1 H – 13 C CP 36. Remarque : Les expériences de CP montrent les signaux provenant des résidus dans une conformation rigide. Étalonnage de l’impulsion et le découplage des paramètres peuvent être directement optimisés sur l’échantillon lorsque la sensibilité est suffisamment élevée pour observer des signaux après moins de 32 ~ scans. Les valeurs d’optimisation initiale sont repris de l’optimisation standard sur un composé de référence. Temps de contact de CP et de niveaux de puissance sont choisis en fonction sur l’intensité du signal maximal. Dans des échantillons de protéine, le temps de contact optimal de CP est généralement entre 200 µs et Mme 2 les paramètres découplage devraient également être réajustés de l’étalonnage sur un composé de référence. Observer attentivement la localisation de la filature des bandes latérales à la fréquence de MAS donnée. Enregistrer un spectre référence 1D 13 C CP qui sert d’empreinte spectrale 1D. Les paramètres typiques sont 128 scans, 1 ms CP temps de contact, 100kHz découplage effectif, un retard de recyclage de 3 s et 25 ms d’acquisition. Calibrer les 1 H et des déplacements chimiques de 13 C suivant l’IUPAC recommandations 37. Déterminer la fréquence de résonance du signal 1 H DSS (déplacement chimique de 0 ppm) ; 13 C les déplacements chimiques sont référencées aux déplacements 1 H (généralement ~ 0.25144, dérivé de la ratio des 13 C le de fréquence de résonance 1 H 37). Processus le 1D 1 H – 13 C CP expérimenter sans apodisation fonctionne (voir les figures 3 a et B pour la détection sur un assemblage de protéines homogène bien ordonné). Choisissez un pic isolé pour estimer la largeur des raies dans l’échantillon, d’homogénéité et d’ordre structurel. Largeurs de typique 13 C pour assemblages de protéine biologique bien ordonnées sous MAS à des champs magnétiques élevés allant de 20-150 Hz (mesurée par la pleine largeur à mi-hauteur (FWHH)). Estimer le ratio signal-bruit (S/N) dans le spectre de la CP. NOTE : Le rapport signal/bruit dépend de nombreux facteurs, dont principalement la quantité d’échantillon dans le rotor, le degré de rigidité de la structure des protéines et la présence d’une seule conformation moléculaire. Comme règle générale, un échantillon doit être approprié pour spectroscopie multidimensionnelle de SSNMR si un signal est observé dans un optimisation 1D 1 H – 13 C CP spectre enregistré avec 64 scans. Résonances zoom dans la région spectrale du carbonyle d’épine dorsale de protéine localisées (principalement) entre 165-180 ppm. Estimer la teneur en structure secondaire dans l’Assemblée par la position des résonances protéine épine dorsale carbonyle avec les résonances de résidus en α-hélicoïdale ou conformation β-brin décalé vers les bas champs ou upfield, respectivement (voir figure 3 b pour une sous-unité principalement α-hélicoïdale) 38. 1 D INEPT Experience mis en place un 1D 1 H – 13 C INEPTE expérience pour sonder les pièces très mobiles (sub-µs) de l’Assemblée de protéine. NOTE : GARP découplage de quelques kHz au cours de l’acquisition de 39 est couramment utilisé, permettant des courts délais interscan (typiquement 1 s) sans endommager la sonde. Enregistrer un spectre INEPTE de référence qui sert d’empreinte pour les segments mobiles protéine ; paramètres typiques sont 128 scans et un temps d’acquisition de 25 m Processus de la 1 H – 13 C INEPTE expérimenter. Le montant ainsi que les positions des signaux sont révélateurs de la quantité de résidus de mobiles et de la composition en acides aminés respectivement. Remarque : Enregistrer également un 2D 1 H – 13 C INEPTE expérience si les signaux sont présents dans le spectre INEPTE 1D (voir la Figure 3 pour obtenir un exemple) pour permettre une identification plus spécifique des acides aminés. En cas de cession ambigu, nous vous recommandons de vérifier les positions de signal de composant de mémoire tampon par solution NMR. Utiliser la base de données BMRB 40 et publié les données 38 pour attribuer les résonances de leur acide aminé correspondant à près de conformation de pelote aléatoire ; le spectre contient uniquement les composants de tampons mobile si non les résidus sont dans un régime très mobile. figure 3 : résultats représentatifs des Acquisition de spectres SSNMR pour un assemblage de protéines bien structuré. A) 13 FID C détectées d’une expérience de polarisation croisée 1 H – 13 C. Expérience de polarisation croisée de 13 C de 1 H – de B). C) 1 H – 13 Experience C INEPTE d’un assemblage de protéines rigide ; uniquement les composants de tampons sont visibles. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure. 3. détermination de structure de l’analyse conformationnelle et 3D cession de résonance séquentielle configurer un 2D 13 C 13 C pilotée par le proton spin diffusion (DESD) 41 l’expérience pour détecter les corrélations intra-résidu 13 C – 13 C, y compris les chaînes latérales. Prendre en charge les valeurs de l’étape de polarisation croisée initiale 1 H – 13 C de la 1D 1 H 13 C CP accède. Définie le temps de mélange à 50 ms pour uniformément 13 échantillons marqués au C et à 100 ms pour sélectivement 13 échantillons marqués au C. Réglez le temps d’acquisition indirecte entre 5-25 ms et l’acquisition directe entre 15 à 25 ms dépendantes de la résolution spectrale intrinsèque, qui peut être estimée par le signal visible dans la longueur de Free Induction Decay (FID) (illustrée dans figure 3 a). Tester plusieurs paramètres de traitement pour trouver des valeurs optimales en ce qui concerne le signal-bruit et résolution dans le spectre, généralement traitement adéquat peut être réalisé en utilisant une fonction de fenêtre qsine avec un déplacement de bell de sinus (SSB) de 2,5-4. Mis en place, enregistrement et processus un 2D 13 C – 13 C DESD avec une expérience de temps mélange intermédiaire pour détecter séquentielle 13 C – 13 C corrélations reliant principalement j’ai – i±1, mais aussi i±2 et i±3 résidus, fonction rigidité épine dorsale de protéine et des éléments de structure secondaire. Le temps de mélange devrait être placé entre 100-200 ms pour les échantillons étiquetés de façon uniforme. Toutes les autres valeurs peuvent être repris depuis le court mélangeant temps 2D 13 C – 13 C DESD. Mis en place, enregistrement et processus un 2D 15 N – 13 C N j’ai CA j’ai et j’ai CO j’ai experiment utilisant un CP 1 H – 15 N et un transfert de 13 C 15 N – CP spécifique 42. optimiser le transfert de polarisation 15 N – 13 C de façon 1D directement sur l’échantillon, basée sur l’intensité maximale dans la région de CA ou de carbonyle, respectivement. Communication graphiqueles valeurs de CAL pour le temps de contact spécifique CP sont entre 2-6 m mis en place, enregistrement et procédé à une série de 2D 15 N-(13 C-) 13 C des expériences dans le but de l’affectation. Remarque : La première valeur du paramètre 15 N – 13 C polarisation transfert seront prélevée le N j’ai CA j’ai / N j’ai CO j’expérimente. Les corrélations intra-résidus sont établies de N j’ai (CA j’ai) CB i et N j’ai (CA j’ai) CO j’ai, en utilisant respectivement rêve 43 et DESD 13 C – 13 C transferts de polarisation. La connectivité séquentielle (i-1) résidus (i) de résidus prélevée sur des N j’ai (CO i-1) CA i-1 et N j’ai (CO i-1) CX i-1 expériences, les deux en utilisant un DESD 13 C – 13 C transfert de polarisation. Programme d’analyse de choisir un NMR comme CcpNmr analyse 44 ou SPARKY 45 Charger les spectres 2D dans le logiciel (exemplifié par analyse CcpNmr) et charger la séquence protéique primaire. Commence par l’identification des types d’acides aminés visibles dans le court-mélange 13 C – 13 spectre C DESD. Connecter les atomes de carbone des systèmes de spin pour permettre le " type de résidus " Attribution spécifique ; Voir Figure 4 a de l’affectation d’un résidu thréonine. Identifier les résidus autant que possible ; Cela dépendra de la taille de sous-unité protéique, la résolution spectrale et la dispersion. Overlay le court-le mélange avec le spectre de la DESD C intermédiaire-mélange 13 C – 13. Remarque : Les sommets supplémentaires visibles dans la mélange intermédiaire DESD découlent principalement de séquentiel (résidu i – résidus i±1) contacts. Un exemple d’une affectation de deux résidus séquentiel est illustré dans la Figure 4 b. Marquer les pics de résonance d’un système de rotation et trouver des corrélations à la DESD intermédiaire-mélange avec des fréquences de résonance des autres systèmes de spins. Dans les petites protéines ou de peptides, il peut être possible d’obtenir la cession de toute résonance séquentielle après des expériences de DESD 2D 13 C – 13 C. Remarque : Dans les résidus de motifs brins β structure secondaire i – résidu i±2 13 C – 13 C contacts pourraient montrent des signaux plus intenses que les contacts séquentiels en raison de leur proximité dans l’espace ; dans les résidus de motifs de structure secondaire α-hélicoïdale, contacts de i – résidu i±3 13 C – 13 C peuvent aussi conduire à signaux intenses. Si intermédiaire-mélange des expériences DESD sélectivement 13 C marqué échantillons sont disponibles, tu les couvriras sur le spectre court-mélange (si disponible sur la sélectivement 13 C marqué échantillon) et assigner le supplemental pics, prenant en compte l’étiquetage sélectif 13 C modèle (1, 3 – 13 C et 2 – 13 C glycérol 32 , 33 ou 1 – 13 C et 2 13 C glucose 29 , 30 , 31. Remarque : par exemple, dans un 2 – 13 C glycérol étiqueté échantillon plusieurs types d’acides aminés sont marqués sur la position de Cα sans les carbones adjacents (Cβ et C ') étiqueté, favorisant donc la Cα-Cα transfert entre les résidus adjacents. Dans les échantillons sélectivement marqués, à longue distance 13 C – 13 C corrélations pourraient s’accumuler déjà dans mélange intermédiaire 13 C – 13 C DESD expériences. Si un pic ne peut s’expliquer par une corrélation séquentielle, il peut survenir d’un contact à longue distance. Pour commandé hautement structures sous-unité homogène dans l’assembly macromoléculaire, qu’un jeu de résonances devrait être visible dans le spectre. Si doublé (ou encore multiplié) résonances sont visibles pour les atomes de 13 C ou protéine épine dorsale s’étire, conformations deux (ou plusieurs) pour l’atome ou de la portion de la séquence primaire à l’Assemblée, il existe respectivement,. Permet d’identifier les fréquences de résonance de 15 N pour chaque résidu le spectre NCA 2D contenant des corrélations de Cα intra-résidu 15 N – 13. Si la résolution dans la dimension de 13 C n’est pas suffisante, utiliser les informations supplémentaires sur la résonance Cβ dans le 2D NCACB d’identifier la fréquence de résonance intra-résidu 15 N. Utiliser les spectres NCACO contenant intra-résidu 15 N – 13 C – 13 C corrélations et 2D NCACB (i) identifier la fréquence de 15 N intra-résidus et (ii) à lever les ambiguïtés dans les 13 C – 13 C DESD avec l’aide de la dimension supplémentaire 15 N. L’inversion du signal en raison du transfert de rêve conduit à l’observation des corrélations de Cα Cβ typiques pour lequel le signal Cα est positif et le signal Cβ négatif. Plus chaîne latérale 13 C, si elle est visible dans le spectre, redeviennent positifs. figure 4 : 2 dimensions 13 C – 13 C SSNMR DESD expérimente sur un bien ordonnées, uniformément 13 C, assemblage de protéines marquées N 15. A) bref temps DESD de mélange (mélange de 50 ms). B) cession du tronçon Ile32 – Thr33 2-résidu à l’aide de la superposition de la courte durée de mélange DESD avec un temps de mélange longtemps DESD (mélange de 200 ms). s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure. détermination de la structure secondaire utiliser 13 Cα, valeurs des déplacements chimiques Cβ 13 pour calculer le produit chimique secondaire changement 46 ΔδCα-ΔδCβ , indicative de la structure secondaire. Calculer 13 Cα(assigned) – 13 Cα (pelote aléatoire) et 13 Cβ(assigned) – 13 Cβ (pelote). Remarque : Les valeurs de déplacement chimique pour les résidus de conformation de pelote aléatoire peuvent provenir de 38. Terrain ΔδCα-ΔδCβ, positive ou négative des valeurs pour > 3 résidus d’affilée indiquent respectivement les brins β ou conformation α-hélicoïdale ; résidus glycine et la proline peuvent afficher les valeurs des déplacements chimiques inhabituels, comme ils agissent souvent comme " briseurs de structure secondaire ". Prédire les angles dièdres de protéine de déplacements chimiques affectées OUC TALOS + 47 , 48 PREDITOR 49 , 50. Les prédit des angles dièdres Phi/Psi reflètent la structure secondaire de la Marie de protéine AnesieNIT et sont utilisés comme structurelle retenue tout au long du processus de modélisation. Collection de contraintes structurelles Set up et enregistrer un 2D 13 C – 13 C DESD expérimenter un temps de mélange depuis longtemps pour détecter à grande distance 13 C – 13 C corrélations. Un mélange typique fois varient de 400 ms à 1 s. utiliser sélectivement 13 échantillons marqués au C, comme la dilution de spin qui améliore grandement le transfert de polarisation entre les carbones lointains. Recouvrement le long-mélange 2D 13 C – 13 C DESD enregistrés sur échantillon sélectivement marqué sur le mélange des intermédiaires 13 C – 13 C DESD, si possible enregistrée sur le même échantillon sélectivement marqué. Pics supplémentaires proviennent des corrélations entre atomes de 13 C plus éloignés. Au cours de l’affectation de résonance, prendre en compte le régime d’étiquetage sélectif. Évaluer attentivement la résolution du spectre afin de définir une fenêtre de tolérance affectation, c’est-à-dire dépendant sur les résonances de résolution spectrale dans une certaine gamme de ppm, que ce qui peut contribuer au signal. L’écart de la précision de cession est calculé automatiquement dans les programmes de cession de NMR comme l’analyse de CcpNmr ou SPARKY. Si un signal peut être expliqué par une séquentielle (résidu i – i±1 de résidus) ou un milieu de gamme 13 C – 13 C contact (résidu i – résidus j’ai ± 2, 3 ou 4), maintenir cette affectation depuis polarisation relayée transfert peut expliquer le même si la distance de 13 C – 13 C est supérieure à la plage de contact visible attendue corrélation. Classer les affectations de longue portée 13 C – 13 C contacts dans les signaux de fréquence sans équivoque et ambiguë. Dans le cas de fréquences autorisées sans ambiguïté, affectation de seul résonance est possible par rapport à la fenêtre de tolérance affectation. NOTE : Affectations ambigues contiennent toutes les affectations de résonance possible dans la fenêtre de tolérance. Les ambiguïtés peuvent être soulevées simultanément lors du calcul de structure par cycles itératifs d’homonymie issu des structures préliminaires calculés en utilisant seulement les dispositifs de retenue non ambiguës, interprété dans les routines de calcul comme ARIA 51 ou 52 de la UNIO. En outre, les données structurelles provenant de différentes sources biophysiques (p. ex. muté ou tronquée des sous-unités de solution NMR ou cristallographie aux rayons x, les mesures de masse-par-longueur, cryo-EM carte) permet également de réduire le niveau de l’ambiguïté, pour exemples, voir 1 , 3 , 53 , , du 54 55 , 56 , 57 , 58. détection des dispositifs de retenue de la distance intermoléculaire dans un assemblage de protéines symétrique mis en place un 2D 15 N – 13 expérience 59 C douleur-CP sur un mixte (50/50) U – 15 N – / U – 13 C marqué sample. Les signaux détectés sur le spectre de la douleur-CP devraient découler de leurs 15 N – 13 C proximités. Utiliser les résonances précédemment assignées pour effectuer l’affectation des contacts intermoléculaires. Créer un 2D 13 C 13 DESD C avec un temps de mélange long (> 600 ms) sur un (50/50) mélangé (1, 3 – 13 C)-/ (2 – 13 C)-glycérol ou (1 – 13 C)-/ (2 – 13 C)-échantillon de glucose. Remarque : Les signaux détectés dans ce spectre de 13 C – 13 C proviennent de moléculaire inter et intra-13 C – 13 C proximités. Cependant, la complémentarité élevée de ces deux régimes d’étiquetage permet de paires une résonance particulière se poser sans ambiguïté de leurs contacts, par exemple, une sérine CA dans le (2 – 13 C)-glucose marqué sous-unités qui sont corrélés à un CB de sérine à le (1 – 13 C)-glucose marqué des sous-unités. Overlay le spectre de l’échantillon mélangé aux spectres 2D 13 C – 13 C enregistré sur homogènement étiqueté échantillons de ((1,3-13C) et (2 – 13 C)-glycérol ou (1 – 13 C)- et (2 – 13 C)-glucose échantillons étiquetés). Les signaux supplémentaires dans le spectre enregistré sur l’échantillon mélangé devraient découler de leurs interactions. Structure de modélisation de données SSNMR préparer les listes de retenue SSNMR nécessaires pour le calcul de structure : séquence de la protéine (1) ; (2) dispositifs de retenue intra moléculaires distance sans ambiguïté ; (3) restrictions de distance ambigu intra moléculaires ; (4) angle de dièdre TALOS-base restrictions ; (5) dispositifs de retenue inter moléculaires distance sans ambiguïté ; (6) restrictions de distance ambigu inter moléculaires ; (7) d’autres données par d’autres techniques biophysiques (p. ex. les paramètres de symétrie des masse-par-longueur,). Plusieurs examens donnent un aperçu de la modélisation de structure basée sur des données SSNMR et en conjonction avec les données structurelles complémentaires 14 , 60 , 15 , 19 , , du 61 62 note que les contraintes de distance ambiguë peuvent être désambiguïsés lors du calcul de structure par rapport à l’enquête préliminaire modèles structurels basés sur les restrictions de distance sans ambiguïté. Pour assurer l’exactitude de la structure, observer attentivement si la structure converge vers un seul pli. figure 5 : Intra – et intermoléculaires contacts dans les assemblys de protéine symétrique. Représentation schématique des intra – et intermoléculaires 13 C – 13 C contacts à longue distance dans un assembly macromoléculaire hélicoïdal. Les sous-unités sont colorées en blanc et rouge pour illustrer l’étiquette mixte des sous-unités ; c.-à-d. avant l’Assemblée, un mélange 1:1 des deux différents régimes d’étiquetage a été réalisé (par exemple 1 – 13 C glucose et 1 – 13 Glucose C). A) intramoléculaire 13 C – 13 C contacts à longue distance (flèche bleue en pointillés) ; B) intermoléculaire 13 C – 13 C contacts à longue distance (rouge déçus des flèches). s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.