Here the results of four replicative multi-gene plasmids for β-carotene (yellow) and lycopene (red) production. One integrative multi-gene plasmid for disrupting the ADE2 locus was constructed, the colonies of which are red.
Cloning CDSs into the entry vector (pYTK001)
ERG20 was amplified from the yeast genome and the three carotenoid genes crtE, crtYB, crtI from the plasmid pLM49448 into the entry vector pYTK001 as described. Yeast promoters pENO2, pTIP1, pPYK1 and pPDC1 and terminators tTDH2, tHSP26, tADH2, tACS2 were cloned as part plasmids. A point mutation was introduced into CrtYB (G247A) for producing lycopene and a BTS1-ERG20 fusion construct was created to better channel the prenyl intermediate to carotenoids. Figures 6A and 6B show a representative plate of the successful cloning of a part plasmid and provide an example of the green/white screening with 90.35 ± 4.22% total colonies being potentially correct (white).
Assembly of transcription units (cassette plasmids)
Before assembling the cassette plasmid, the design of the multi-gene plasmid was finalized. For the four carotenoid TUs, four intermediate vectors with different connectors were cloned first following step 3.1. Figures 6C and 6D show a representative plate from a successful assembly of the intermediate vector and provide an example of the red/green screening, with 17.56 ± 3.32% total colonies being positive (green). Although this ratio is relatively low, the screening is greatly facilitated by the green fluorescence.
Next, the TUs for BTS1-ERG20, ERG20, crtE, crtYB, crtYB(G247A), and crtI were assembled following step 3.2 (Table 1). Figures 6E and 6F show a representative plate of a successful assembly of the TUs and provide an example of a green/white screening method, with 65.02 ± 4.99% total colonies being positive (white).
Assembly of multi-gene plasmids
Four replicative and one integrative multi-gene plasmids were assembled. For replicative multi-gene plasmids, ERG20, BTS1-ERG20, crtE, crtYB, crtYB (G247A) and crtI were assembled in four different combinations, creating four plasmids: two for β-carotene and two for lycopene production (Table 2). The ratio of potentially correct colonies (green) for the intermediate vector cloning was 1.83 ± 0.15% (Figures 7A and 7B). Although this number seems low, the screening was made easy by detecting the green fluorescence (Figure 7B). Once the intermediate plasmid was cloned, the success rate of assembling multi-gene plasmids (white) from the intermediate was 93.77± 1.65% (Figures 7C and 7D). Figures 7E and 7F show a suboptimal assembly of multi-gene plasmids, as the numbers of positive colonies (white) were negligible. After transforming into yeast, colonies producing β-carotene (yellow) and lycopene (red) grew on day three. Four colonies from each plate were streaked out onto fresh plates and grown for two more days. Figures 8A and 8B show that fusing the BTS1-ERG20 directs more geranylgeranyl-pyrophosphate toward the carotenoid production, as seen by darker colors than using the ERG20 alone. Upon extraction49 and quantification of the carotenoids by UV-Vis spectrophotometry with authentic standards, it is seen that fusion of BTS1-ERG20 leads to the production of 0.729 µg/mg β-carotene, which is ~35 fold higher than 0.021 μg/mg β-carotene produced by the strain with ERG20 alone. Likewise, the production of lycopene is ~16.5 fold higher in the strain with BTS1-ERG20 (1.126 μg/mg) compared to ERG20 (0.068 μg/mg) alone (Figure 8C).
Replicative multi-gene plasmids transformed into yeast with (A) BTS1-ERG20 fusion TU and (B) ERG20 TU. On each plate, yeast on the left side has plasmids containing crtE TU, crtYB TU and crtI TU for the production of β–carotene and yeast on the right side has plasmids containing crtE TU, crtYB (G247A)TU, and crtI TU for the production of lycopene.
For the integrative plasmid, the ConLS', sfGFP dropout, ConRE', HIS3 (yeast selection marker), ADE2 5' and 3' homology arms were assembled following step 4.1.2. The 5' and 3' homology arms were 500 bp apart, deleting ~180 amino acids after integration. Additionally, the 5' homology arm had six stop codons towards its 3' end. The mutated ADE2 resulted in red colonies50. The ADE2 gRNA 5'-ATTGGGAC GTATGATTGTTGAGG-3'51 were used and followed step 5.1 for genomic integration. After 3-4 days, red colonies were observed on the YPD plate + 100 μg/mL nourseothricin, indicating that ADE2 had been successfully disrupted (Figures 8D and 8E).
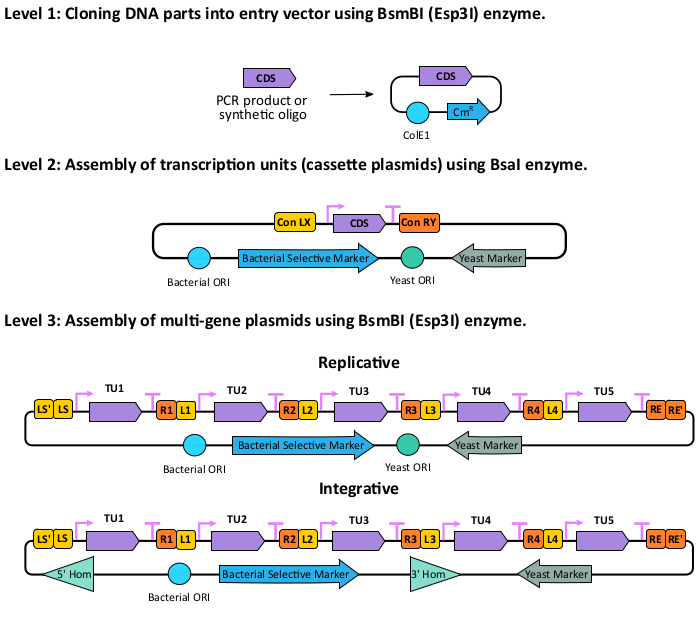
Figure 1: Overview of multi-gene assembly. Assembly takes place in three levels. In0 level 1, the CDS, or any other part, is amplified from the genome or synthesized, and then cloned into the pYTK001 entry vector using the BsmBI (or Esp3I) enzyme. ColE1: E. coli origin of replication; CmR: Chloramphenicol resistant gene. In level 2, the transcription unit (TU) containing a promoter, a CDS, and a terminator is assembled using the BsaI enzyme. In level 3, up to five transcription units are assembled into a multi-gene plasmid using the BsmBI (or Esp3I) enzyme. The multi-gene plasmid can be either replicative or integrative. Please click here to view a larger version of this figure.
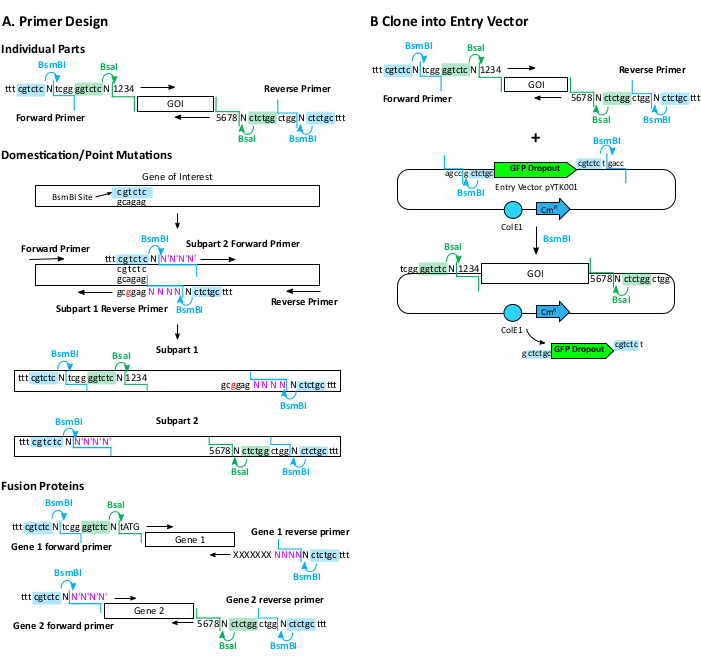
Figure 2: Primer design and cloning of part plasmids. (A) Primer design for amplifying individual parts, domesticating genes or creating point mutations, and assembling fusion proteins. Primers include BsmBI and BsaI recognition and cut sites and the MoClo overhang for proper assembly. MoClo overhangs are represented as 1, 2, 3, 4, and 5, 6, 7, 8. Internal primers for domestication or creating fusion protein contains the BsmBI but not the BsaI sites. The overhangs for these are customized internal sequences (NNNN and N'N'N'N' in purple). Terminal "ttt" are included for optimal enzyme digestion. GOI: gene of interest. (B) Cloning amplified parts into the pYTK001 entry vector using BsmBI (or Esp3I). Complementary overhangs lead to the integration of the part and the removal of the BsmBI recognition site. ColE1: E. coli origin of replication; CmR: Chloramphenicol resistant gene. Please click here to view a larger version of this figure.
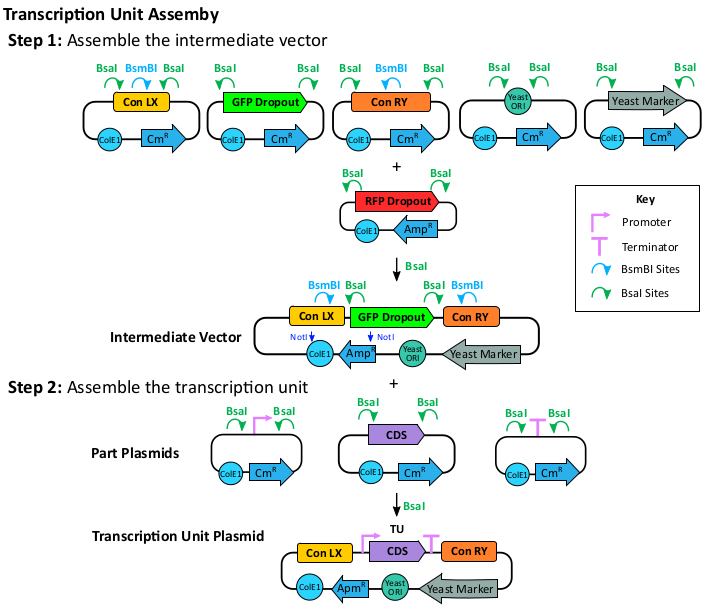
Figure 3: Transcription unit assembly. To assemble the TU plasmids, the assembly of an intermediate vector is recommended to facilitate combinatorial TU assembly. To assemble the intermediate vector, clone the Con LX (X: one of the five left connectors), the sfGFP dropout, the Con RY (Y: one of the five right connectors), a yeast ORI (origin of replication), and a yeast marker part into the mRFP1 dropout vector using the BsaI enzyme. The intermediate plasmid is resistant to ampicillin. The BsaI recognition sites are retained for TU plasmid cloning. To clone the TU plasmid, a promoter, a CDS, and a terminator are assembled into the intermediate vector using BsaI. The cloned TU will have BsmBI sites at the ConLX and ConRY regions for the next step multi-gene assembly. The cloned TU is also resistant to ampicillin. Please click here to view a larger version of this figure.
| Name | ConLX (left connector) | Promoter | CDS | Terminator | ConRY (right connector) |
| BTS/ERG20 TU | LS | pENO2 | BTS1/ERG20 | tTDH2 | R1 |
| ERG20 TU | LS | pENO2 | ERG20 | tTDH2 | R1 |
| crtE TU | L1 | pTIP1 | crtE | tHSP26 | R2 |
| crtYB TU | L2 | pPDC1 | crtYB | tADH2 | R3 |
| crtYBG247A TU | L2 | pPDC1 | crtYBG247A | tADH2 | R3 |
| crtI TU | L3 | pPYK1 | crtI | tACS2 | RE |
Table 1: Transcription units used in this study. Promoters and terminators were amplified from S. cerevisiae. BTS1 (geranylgeranyl diphosphate synthase) and ERG20 (farnesyl pyrophosphate synthetase) were amplified from S. cerevisiae. The genes crtE (geranylgeranyl diphosphate synthase), crtYB (bifunctional lycopene cyclase/phytoene synthase), and crtI (phytoene desaturase) were from Xanthophyllomyces dendrorhous.
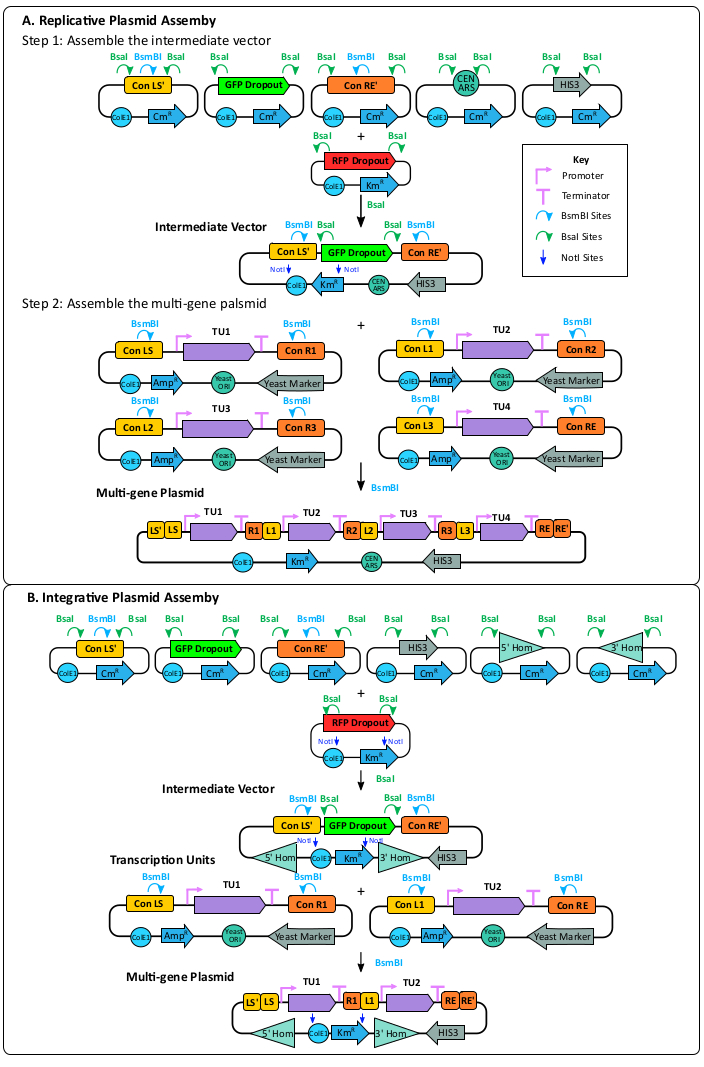
Figure 4: Multi-gene plasmid assembly. (A) Replicative plasmid assembly. Assembly of the replicative intermediate vector includes cloning the Con LS', the sfGFP dropout, the Con RE', a yeast ORI, a yeast marker, and an E. coli origin and marker on the mRFP1 dropout vector using the BsaI enzyme. The ConLS' and ConRE' sites introduce BsmBI recognition sites to the vector. Potentially correct assemblies can be screened by looking for green colonies on a selective plate with kanamycin. The previously assembled TUs can then be cloned into the intermediate vector using the BsmBI enzyme. This plasmid contains a yeast ORI allowing it to replicate in a yeast host. (B) Integrative plasmid assembly. Assembly of the integrative intermediate vector includes cloning the Con LS', the sfGFP dropout, the Con RE', a 5' homology arm, a 3' homology arm, a yeast marker, and the E. coli origin and marker into the RFP dropout vector using the BsaI enzyme. Correct assemblies should appear green on a selective plate with kanamycin. Transcription units previously made can be cloned into the replicative intermediate vector using the BsmBI enzyme. This vector does not have a yeast ORI and will be integrated into the target locus through CRISPR and homologous recombination. Please click here to view a larger version of this figure.
| Name | TU1 | TU2 | TU3 | TU4 | Product |
| B/E-β-Carotene | BTS1/ERG20 | crtE | crtYB | crtI | β-carotene |
| B/E-Lycopene | BTS1/ERG20 | crtE | crtYBG247A | crtI | Lycopene |
| E-β-Carotene | ERG20 | crtE | crtYB | crtI | β-carotene |
| E-Lycopene | ERG20 | crtE | crtYBG247A | crtI | Lycopene |
Table 2: Multi-gene Plasmids used in this study.
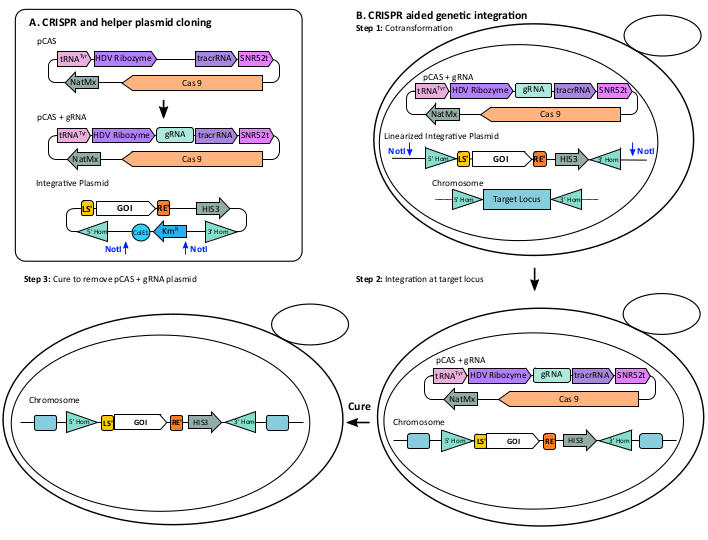
Figure 5: CRISPR integration. (A) CRISPR and helper plasmid cloning. The pCAS plasmid contains the Cas9 endonuclease and components (tRNA promoter, SNR52 terminator, HDV ribozyme, and tracrRNA) for optimal expression of a gRNA. Clone the pCAS+gRNA plasmid by assembling the synthetic gRNA with the linearized pCAS using Gibson cloning. (B) CRISPR aided genetic integration. Step 1: Cotransformation: pCAS +gRNA was co-transformed into yeast with the integrative plasmid containing the gene(s) of interest (GOI), a yeast selective marker, and 5' and 3' homology region targeting the genomic locus. For optimal integration, linearize the integrative plasmid with NotI. Step 2: Integration at target locus: Growing the transformed yeast on a plate selective for the yeast marker, either antibiotic or auxotrophic. Perform genotyping to confirm the integration. Step 3: Cure the pCAS + gRNA plasmid by streaking yeast on a non-selective plate. Please click here to view a larger version of this figure.
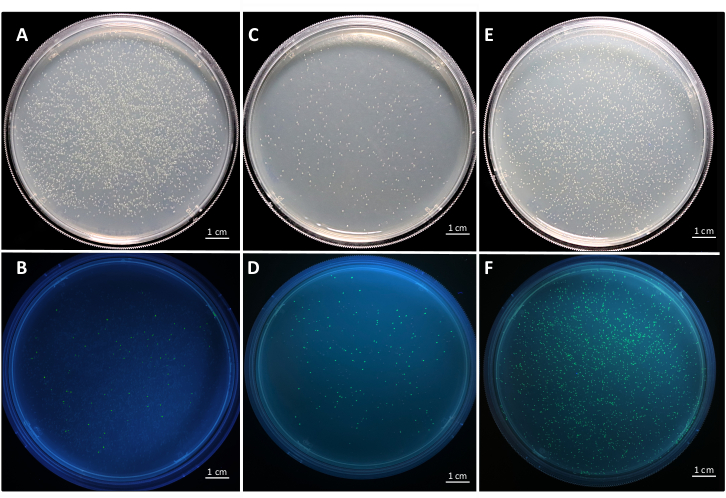
Figure 6: Representative plates of entry vector and transcription unit level cloning in E. coli. Representative plates of the successful cloning of a gene into the entry vector pYTK001 under (A) visible light and (B) UV light. Positive colonies are white and negative colonies are green. The ratio of positive colonies overall colonies is 90.35 ± 4.22 %. Successful assembly of the intermediate vector for transcription unit level assembly and green/red selection under (C) visible light and (D) UV light. Positive colonies are green. The ratio of positive colonies overall colonies is 17.56 ± 3.32 %. Successful assembly of transcription unit from the intermediate vector and green/white screening under (E) visible light and (F) UV light. Positive colonies are white and negative colonies are green.The ratio of positive colonies overall colonies is: 65.02 ± 3.32 %. Data are from three biological replicates. Please click here to view a larger version of this figure.
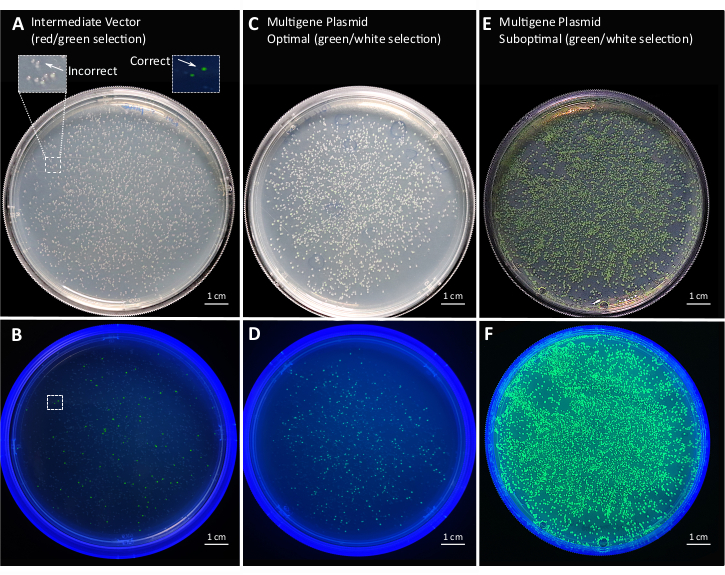
Figure 7: Representative plates of multi-gene plasmid cloning in E. coli. Assembly of the intermediate vector for multi-gene level assembly and red/green selection under (A) visible light and (B) UV light. Positive colonies are green and negative colonies are red. The ratio of positive colonies overall colonies is 1.83 ± 0.15%. Successful assembly of multiple TUs from the intermediate vector and green/white selection (C) under visible light and (D) UV light. Positive colonies are white. The ratio of positive colonies overall colonies is 93.77 ± 1.65%. Suboptimal assembly of multi-gene plasmid under (E) visible light and (F) UV light. The number of positive colonies is negligible. Data are from three biological replicates. Please click here to view a larger version of this figure.
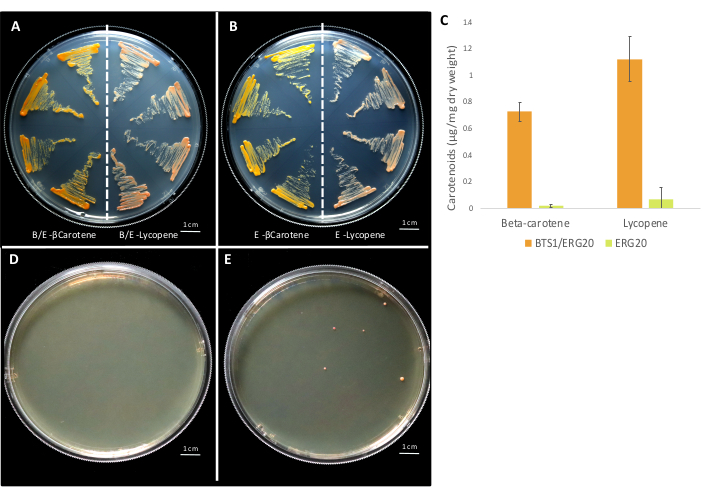
Figure 8: Representative plates of integrative and replicative plasmids in yeast. Replicative multi-gene plasmids transformed into yeast with (A) BTS1-ERG20 fusion TU and (B) ERG20 TU. On each plate, yeast on the left side has plasmids containing crtE TU, crtYB TU and crtI TU for the production of β–carotene and yeast on the right side has plasmids containing crtE TU, crtYB (G247A)TU, and crtI TU for the production of lycopene. (C) quantifying β-carotene and lycopene from yeast extract with four multi-gene plasmids using a UV-vis spectrometer. The maximal absorbance was recorded at 450 nm and 470 nm for β-carotene and lycopene respectively. The absolute quantification was performed using authentic standards (Supplementary Figure S3). Fusion of BTS1-ERG20 leads to the production of ~35-fold higher β-carotene and ~16.5-fold higher lycopene compared to ERG20 alone. Representative plates for the disruption of the ADE2 locus by the integration of a multi-gene integrative plasmid with a gRNA and no helper DNA (D) and with gRNA and a multi-gene integrative plasmid as helper DNA (E). Please click here to view a larger version of this figure.
Supplementary Materials. Please click here to download this file.
