


Summary
Este protocolo describe un método no invasivo para identificar eficientemente las células de fase S para estudios de microscopía aguas abajo, como la medición del reclutamiento de proteínas de reparación del ADN mediante microirradiación láser.
Abstract
La reparación del daño en el ADN mantiene la integridad genética de las células en un entorno altamente reactivo. Las células pueden acumular varios tipos de daño en el ADN debido a fuentes endógenas y exógenas, como las actividades metabólicas o la radiación UV. Sin la reparación del ADN, el código genético de la célula se ve comprometido, socavando las estructuras y funciones de las proteínas y potencialmente causando enfermedades.
Comprender la dinámica espaciotemporal de las diferentes vías de reparación del ADN en varias fases del ciclo celular es crucial en el campo de la reparación del daño del ADN. Las técnicas actuales de microscopía fluorescente proporcionan excelentes herramientas para medir la cinética de reclutamiento de diferentes proteínas de reparación después de la inducción del daño al ADN. La síntesis de ADN durante la fase S del ciclo celular es un punto peculiar en el destino celular con respecto a la reparación del ADN. Proporciona una ventana única para detectar errores en todo el genoma. Al mismo tiempo, los errores de síntesis de ADN también representan una amenaza para la integridad del ADN que no se encuentra en las células que no se dividen. Por lo tanto, los procesos de reparación del ADN difieren significativamente en la fase S en comparación con otras fases del ciclo celular, y esas diferencias son poco conocidas.
El siguiente protocolo describe la preparación de líneas celulares y la medición de la dinámica de las proteínas de reparación del ADN en fase S en sitios de daño del ADN inducidos localmente, utilizando un microscopio confocal de escaneo láser equipado con una línea láser de 405 nm. El PCNA etiquetado (con mPlum) se utiliza como un marcador del ciclo celular combinado con una proteína de reparación de interés marcada con AcGFP (es decir, EXO1b) para medir el reclutamiento de daño en el ADN en la fase S.
Introduction
Varias vías de reparación del ADN han evolucionado para abordar los diferentes tipos de lesiones de ADN que pueden surgir en las células, todas las cuales están altamente reguladas tanto en el espacio como en el tiempo. Uno de los períodos más vulnerables del ciclo celular es la fase S, cuando se produce la síntesis de ADN. Si bien la proliferación es fundamental para la vida, también supone un reto importante. Las células necesitan asegurar una replicación fiel de su genoma para evitar que las mutaciones se transmitan a las generaciones futuras. En consecuencia, la proliferación proporciona un punto terapéutico de intervención que se ha empleado para el desarrollo de enfoques terapéuticos en el campo de la oncología.
Todas las principales técnicas utilizadas para estudiar el reclutamiento de proteínas en las lesiones de ADN tienen sus fortalezas y limitaciones. La microirradiación tiene una mejor resolución espacial y temporal1 que la mayoría de los métodos alternativos, como las imágenes inmunofluorescentes de focos inducidos por radiación ionizante (IRIF), la inmunoprecipitación de cromatina (ChIP) o el fraccionamiento bioquímico. Sin embargo, la micro-irradiación laca la solidez de las técnicas antes mencionadas que pueden muestrear un gran número de células al mismo tiempo.
Para investigar la reparación del ADN en la fase S, uno debe ser capaz de distinguir las células de la fase S en una población de cultivo celular asíncrono. Hay muchos métodos bien conocidos para abordar esto, que involucran la sincronización de las células o la visualización de las diferentes fases del ciclo celular. Sin embargo, ambos enfoques introducen desafíos significativos y posibles artefactos. Los métodos de sincronización química ampliamente utilizados para enriquecer las células en la fase S temprana (por ejemplo, doble bloqueo de timidina, afinicicolina y tratamiento con hidroxiurea) logran la sincronización a través de la inducción del estrés de replicación y, finalmente, el daño del ADN en sí. Esto limita el uso de estos métodos para estudiar los procesos de reparación del ADN en la fase S2. La sincronización a través de la inanición y liberación sérica solo es aplicable a un número limitado de líneas celulares, excluyendo en gran medida las líneas celulares cancerosas que dependen menos de los factores de crecimiento para la progresión del ciclo celular en comparación con las líneas celulares no transformadas. El sistema Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI) es una herramienta particularmente útil para estudiar el ciclo celular, pero tiene una limitación fundamental a la hora de diferenciar entre las fases del ciclo celular S y G23.
Aquí se muestra que el uso de PCNA marcado fluorescentemente como un marcador no invasivo para la fase S limita los inconvenientes de los métodos químicos de sincronización del ciclo celular, al tiempo que permite una mayor especificidad y flexibilidad que el sistema FUCCI. Como marcador único, el PCNA no solo puede resaltar las células de fase S en una población asíncrona, sino que también puede mostrar la progresión exacta de las células dentro de la fase S (es decir, fase S temprana, media o tardía)4. Los bajos niveles de expresión de PCNA exógeno y etiquetado aseguran una interferencia mínima tanto con la progresión del ciclo celular como con los procesos de reparación del ADN. Es importante destacar que el PCNA también sirve como un control interno para la inducción adecuada del daño en el ADN, ya que está involucrado en la reparación de varias lesiones de ADN y se recluta en sitios de daño al ADN inducidos localmente1,4.
Los experimentos presentados aquí demuestran cómo medir la dinámica de reclutamiento de EXO1b en fase S y cómo esto se ve afectado por el inhibidor de PARP bien establecido, olaparib. La actividad de la nucleasa EXO1b es relevante para una amplia gama de vías de reparación del ADN, incluida la reparación de desajustes (MMR), la reparación por escisión de nucleótidos (NER) y la reparación de roturas de doble cadena (DSB). En la fase S, EXO1b juega un papel importante en la recombinación homóloga (HR) a través de la formación de voladizos de 3' ssDNA durante la resección del ADN5. EXO1b se ha implicado aún más en la replicación del ADN con funciones en la activación del punto de control para reiniciar las horquillas de ADN estancadas, así como la eliminación del cebador y la maduración del fragmento de Okazaki en la hebra rezagada durante el desplazamiento de la hebra en la replicación5. El reclutamiento de EXO1b a sitios de ADN dañados está regulado por la interacción directa con poli (ADP-ribosa) (PAR)6,7. Debido a las numerosas implicaciones específicas del ciclo celular de EXO1b, es una excelente opción para los estudios de reclutamiento específicos de fase S que utilizan PCNA.
Protocol
1. Cultivo de células derivadas del osteosarcoma humano (OS U-2)
NOTA: Las células del sistema operativo U-2 son ideales para estos estudios, ya que tienen una morfología plana, un núcleo grande y se adhieren fuertemente a varias superficies, incluido el vidrio. También se podrían utilizar otras líneas celulares con características similares.
- Para el cultivo de líneas celulares de SO U-2, use el medio 5A de McCoy suplementado con suero bovino fetal (FBS) al 10% y antibióticos (100 U/ ml de penicilina y 100 μg / ml de estreptomicina). Incubar células a 37 °C en una atmósfera humidificada que contenga 5% de CO2. Para los estudios de microscopía, mantenga el cultivo celular en un plato de 10 cm para proporcionar un recuento celular suficiente.
- Cuando las células se acercan al 90% de confluencia (~ 7 x 106 células / plato de 10 cm), divida las células.
- Enjuague las células con PBS para eliminar los inhibidores de tripsina contenidos en el suero.
- Agregue 1 ml de tripsina-EDTA y asegúrese de que la capa celular esté igualmente cubierta.
- Incubar a 37 °C hasta que la capa celular se levante de la placa (aproximadamente 6 min).
- Resuspend las células tripsinizadas en suero que contienen medios para inactivar la tripsina y agregar 1/10del volumen (~ 0.7 x 106 células) en una nueva placa de 10 cm que contiene 10 ml de medio de crecimiento suplementado.
- Antes de la experimentación, pruebe rutinariamente las células para detectar la contaminación por micoplasma utilizando el kit de detección universal de micoplasma siguiendo la recomendación del fabricante.
2. Infección retroviral
NOTA: Para conocer las medidas de seguridad de BSL-2 y mientras se trabaja con virus recombinantes, consulte: Directrices de los NIH, Sección III-D-3: Virus recombinantes en cultivo de tejidos.
- Seque 4 x 106 células HEK293T para lograr ~ 60% de confluencia dentro de las 24 h después de emplatar en un plato de cultivo de 10 cm.
- Para cultivar HEK293T, siga los pasos de cultivo del sistema operativo U-2 descritos en 1.1-1.3 de este protocolo. Para HEK293T sustituya el medio 5A de McCoy por DMEM. Asegúrese de lavar siempre suavemente las células HEK293T a medida que se adhieren débilmente a las placas de cultivo de tejidos.
- Transfectar células HEK293T utilizando un reactivo de transfección a base de lípidos para el envasado viral de plásmidos.
- Para vectores retrovirales, combine 1,5 μg de VSV-G (Addgene #8454) y 1,5 μg de vectores de empaquetamiento pUMVC (Addgene #8449) junto con 3 μg del vector que contiene el gen de interés (en una columna vertebral de vector retroviral con resistencia a la puromicina) en 250 μL de medios séricos reducidos Opti-MEM en un tubo de microcentrífuga. Añadir 1 μL de reactivo P3000 por cada μg de ADN añadido a la mezcla Opti-MEM/DNA (en este caso 6 μL) y mezclar suavemente mediante golpeteo. No vórtice ni pipetee hacia arriba y hacia abajo.
- En otro tubo de microcentrífuga, combinar 2 μL por μg de ADN (en este caso 12 μL) de reactivo de transfección con 250 μL de medios séricos reducidos Opti-MEM.
- Combine las dos mezclas (500 μL combinados, no vórtice, solo mezcle golpeando suavemente) y deje que se incube durante 15 minutos a temperatura ambiente.
- Con cuidado, agregue la mezcla gota a gota a las células HEK293T semilladas sin separar las células. Gire las placas suavemente.
- Infección viral para generar líneas celulares estables.
- Retire el virus que contiene sobrenadante de las células HEK293T 72 h después de la transfección. Filtre cuidadosamente la solución con un filtro de 0,45 μm para eliminar los restos celulares y las células desprendidas. Opcionalmente, agregue 8 μg / ml de polibreno al sobrenadante viral para facilitar la infección viral.
- Agregue el virus que contiene sobrenadante a las células del sistema operativo U-2 a ~ 50% de confluencia en un plato de 10 cm (~ 3 x 106 células). Siembra las células del sistema operativo U-2 el día anterior.
- Infectar durante 6-16 h antes de retirar y desechar el sobrenadante que contiene el virus.
NOTA: Para lograr la cantidad deseada de sobreexpresión para el gen de interés, incubar una serie de diluciones virales durante un período de tiempo fijo. Compruebe los niveles de expresión del transgén en cada línea celular recién establecida con Western Blot comparándolo con los niveles endógenos. - Permita que las células seleccionen en presencia de antibióticos apropiados (durante 3-4 días en caso de puromicina a una concentración final de 2 μg / ml) y verifique la expresión de la proteína fluorescente marcada como gen de interés bajo un microscopio.
- Repita estos pasos para generar líneas celulares con doble etiqueta. En los experimentos presentados aquí mPlum-PCNA se expresó a partir de un vector retroviral (pBABE) combinado con EXO1B-AcGFP, también expresado a partir de un vector retroviral (pRetroQ-AcGFP1-N1).
3. Preparación de células para la microirradia
- Células de revestimiento: 24 h antes del experimento, coloca un total de 8,0 x 104 células en un volumen entre 500 μL-1 mL de medios (para aproximadamente el 70% de confluencia) en una cubierta de cuatro pomos con cámara con un fondo de vidrio de borosilicato No. 1.5 que ofrece resultados ideales para microscopía confocal de alto aumento y microirradiación láser. Una mayor confluencia celular permite medir más células en un solo campo de visión (FOV); sin embargo, las diapositivas totalmente confluentes introducirán irregularidades en el ciclo celular.
- Medios de imagen: Una hora antes de la microirradiación, intercambie un medio de crecimiento regular para FluoroBrite DMEM suplementado con 10% de FBS, 100 U/mL de penicilina y 100 μg/mL de estreptomicina, 15 mM de HEPES (pH=7,4) y 1 mM de sodio-piruvato. Este medio de imagen ayuda a maximizar la relación señal-ruido permitiendo la detección de fluorescencia muy tenue. Dado que contiene HEPES, también estabiliza el pH en ausencia de una atmósfera de CO2 al 5%.
- Aplique cualquier tratamiento adicional antes de la obtención de imágenes en este paso. En los experimentos aquí presentados, las células fueron pretratadas una hora antes de la obtención de imágenes con olaparib (inhibidor de PARP, a 1 μM de concentración final) o un vehículo de control (DMSO)1,8,9.
4. Preparación del microscopio y selección de células de fase S para la obtención de imágenes.
- Utilice un sistema confocal que tenga las propiedades similares al sistema descrito aquí para obtener los mejores resultados. Los experimentos aquí presentados se realizaron utilizando un microscopio confocal montado en un soporte de microscopio invertido (ver Tabla de Materiales).
NOTA: El microscopio utilizado aquí estaba equipado con un módulo láser FRAP de 50 mW y 405 nm y un objetivo de apocromato de plan de aceite de 1,4 NA de 60x. El cabezal de escaneo confocal tenía dos opciones de escáner: un escáner galvano (para alta resolución) y un escáner resonante (para imágenes de alta velocidad).- Introduzca la recuperación de fluorescencia después del láser de fotobleaching (FRAP) en la muestra a través de un dispositivo de galvano XY controlado por software. Utilice una línea láser de 488 nm para excitar AcGFP y una línea láser de 561 nm o 594 nm para excitar mPlum.
NOTA: La siguiente combinación de filtros da resultados óptimos: utilizando un filtro de paso largo de 560 nm, la luz de emisión con una longitud de onda inferior a 560 nm se pasó a través de un filtro de emisión de 525/50 nm para AcGFP, mientras que la luz de emisión con una longitud de onda superior a 560 nm se pasó a través de un filtro de emisión de 595/50 nm para mPlum. Se podría utilizar cualquier conjunto de filtros apropiado (por ejemplo, FITC / TRITC, GFP / mCherry, FITC / TxRed) que garantice un sangrado de fluorescencia mínimo.
- Introduzca la recuperación de fluorescencia después del láser de fotobleaching (FRAP) en la muestra a través de un dispositivo de galvano XY controlado por software. Utilice una línea láser de 488 nm para excitar AcGFP y una línea láser de 561 nm o 594 nm para excitar mPlum.
- Encienda la cámara ambiental y los componentes del microscopio.
- Encienda la calefacción (etapa, objetivo y cámara ambiental cuando sea posible), el suministro de CO2 y el regulador de humedad al menos 4 h antes del inicio del experimento para garantizar el equilibrio térmico para una adquisición de imagen estable.
- Inicialice las fuentes de luz junto con las líneas láser al menos 1 h antes de la transferencia de las células al microscopio.
- Seleccione células de fase S en una población asíncrona utilizando PCNA marcado fluorescentemente como marcador. Haga esto siguiendo los pasos a continuación.
- Busque el patrón de localización único del PCNA marcado con mPlum en fase S que haga posible la identificación de esta fase del ciclo celular. El PCNA tiene una distribución completamente homogénea en el núcleo en las fases G1 y G2 del ciclo celular, a la vez que se excluye de los nucleolos. En la fase S, el PCNA forma focos en la ubicación de los replisomas en el núcleo. La Figura 1 muestra los diferentes patrones de focos de PCNA a lo largo de la fase S, lo que permite incluso diferenciar la fase S temprana, media y tardía.
- Mire a través del ocular para seleccionar un FOV que tenga suficientes células de fase S para la microirradia. Las células asincrónicas del sistema operativo U-2 generalmente tienen un 30-40% de su población en fase S.
- Trate de evitar los niveles extremos en los niveles de expresión (células brillantes y tenues por igual) tanto para el PCNA como para la proteína de interés (POI), en este caso EXO1b-AcGFP, lo que podría conducir a artefactos experimentales.
- Al encontrar un FOV adecuado, trate de evitar escanear el campo durante mucho tiempo para minimizar el fotobleaching y el daño no deseado del ADN.
- Establezca la región de interés (ROI) deseada para la microirradia. Utilizando el software asociado (consulte tabla de materiales),establezca el ROI deseado insertando primero líneas binarias (establezca el número deseado de líneas y espaciado). Haga clic en Binario y,a continuación, haga clic en Insertar línea | Círculo | Elipse para dibujar el número deseado de líneas.
- Convierta estas líneas binarias en ROI y finalmente convierta estos ROI en ROI de estimulación. Para hacer eso, primero haga clic en ROI, luego haga clic en Mover binario a ROI, luego haga clic derecho en cualquiera de los ROI y seleccione Usar como ROI de estimulación: S1. Coloque estas líneas en el FOV para pasar a través del núcleo de las células. Se utilizaron ROI con una longitud de 1024 píxeles que abarcaban todo el campo de visión en todo el protocolo.
5. Microirradiación para tinción de inmunofluorescencia o imágenes de lapso de tiempo.
- Determinación de los ajustes óptimos de microirradiación.
- Antes de la microirradiatura de las células, tome una imagen de mayor resolución del FOV para identificar los focos de PCNA para su posterior análisis. En lugar de escaneo secuencial, registre simultáneamente los dos canales ópticos utilizados (verde y rojo), para evitar el movimiento celular entre el escaneo en las dos longitudes de onda. Para una resolución adecuada de los focos, use al menos una resolución de campo de 1024 x 1024 píxeles con zoom de 1x (tamaño de píxel de 0.29 μm en el sistema de imágenes utilizado aquí), con una velocidad de escaneo de 1/8 de fotograma / s (4.85 μs / píxel) con un promedio de 2x. Una vez que estos parámetros estén configurados en las ventanas A1 LFOV Compact GUI y A1 LFOV Scan Area, presione el botón Capturar para grabar el FOV.
NOTA: Es importante mantener el mismo tamaño de píxel a lo largo de los experimentos para garantizar resultados comparables. - Para configurar la microirradiación, abra la pestaña Estimulación ND en el software de imágenes para acceder a la ventana Programación de tiempo (A1 LFOV / Galvano Device). Esto utiliza los escáneres de galvano para adquirir una serie de imágenes de preestimulación, estimular (utilizando el láser LUN-F 50 mW 405 nm FRAP), y luego adquirir una serie de imágenes de post-estimulación nuevamente utilizando los escáneres de galvano. Primero configure tres fases en la ventana Programación de tiempo. En la columna Acq/Stim, seleccione Adquisición | | blanqueador Adquisición para las tres fases respectivamente. Para la fase de blanqueo, establezca S1 como el ROI.
NOTA: En el experimento presentado aquí, no se adquirieron imágenes durante la fase de estimulación. - En la ventana Galvano XY,configure los factores clave para la microirradiación: salida de potencia láser de 405 nm, tiempo de permanencia (la iteración es 1 por defecto en este sistema). En los experimentos presentados aquí, las células fueron irradiadas con el láser FRAP de 405 nm (50 mW en la punta de la fibra) al 100% de potencia de salida con un tiempo de permanencia de 1000-3000 μs.
NOTA: Debido a que el tiempo de permanencia del láser es por píxel, siempre que el tamaño del píxel siga siendo el mismo, la relación entre el tiempo de permanencia y la densidad de potencia será comparable entre los diferentes FOV. La Figura 2A muestra el uso de proteínas específicas de la vía de respuesta al daño del ADN (DDR) (FBXL10 para DSB y NTHL1 para el daño de la base oxidativa) para optimizar la configuración de potencia del láser para la inducción de daños específicos. Estas líneas celulares estables se generaron con infección viral siguiendo la sección 2 del protocolo.
- Antes de la microirradiatura de las células, tome una imagen de mayor resolución del FOV para identificar los focos de PCNA para su posterior análisis. En lugar de escaneo secuencial, registre simultáneamente los dos canales ópticos utilizados (verde y rojo), para evitar el movimiento celular entre el escaneo en las dos longitudes de onda. Para una resolución adecuada de los focos, use al menos una resolución de campo de 1024 x 1024 píxeles con zoom de 1x (tamaño de píxel de 0.29 μm en el sistema de imágenes utilizado aquí), con una velocidad de escaneo de 1/8 de fotograma / s (4.85 μs / píxel) con un promedio de 2x. Una vez que estos parámetros estén configurados en las ventanas A1 LFOV Compact GUI y A1 LFOV Scan Area, presione el botón Capturar para grabar el FOV.
- Imágenes de lapso de tiempo.
- Configure las imágenes de lapso de tiempo para la ventana de tiempo y los intervalos deseados utilizando la programación de tiempo, la GUI compacta de A1 LFOV y las ventanas de área de escaneo de LFOV A1. En los experimentos presentados aquí, el reclutamiento de EXO1b y PCNA se fotografió durante 12 minutos, escaneando el FOV cada 5 segundos, a 1024 x 1024 píxeles / campo, utilizando un zoom de 1x (lo que resultó en un tamaño de píxel de 0.29 μm en el sistema de imágenes utilizado aquí) con una velocidad de escaneo de 0.35 cuadros / s (1.45 μs / píxel) sin promediar para reducir el blanqueamiento fotográfico.
- Optimice los ajustes de potencia del láser %, ganancia y desplazamiento para reducir el fotolcolorado durante la imagen en la ventana A1 LFOV Compact GUI. Si uno tiene como objetivo medir tanto POI como PCNA, use escaneo simultáneo en lugar de escaneo secuencial para evitar el movimiento celular entre el escaneo del campo en busca de los dos fluoróforos separados.
- El sistema de imágenes se utilizó con los siguientes ajustes. Para la línea láser de 488 nm (20 mW): 7% de potencia láser, ganancia: 45 (detector GaAsP) con y desplazamiento de 2, para la línea láser de 561 nm (20 mW): 4% de potencia láser, ganancia 40 (detector GaAsP) con y desplazamiento de 2.
- Dependiendo de la cinética de la proteína, extender o acortar el intervalo entre imágenes o la duración del lapso de tiempo total. En la ventana Programación de tiempo, establezca el intervalo y la duración deseados para la fila Adquisición de la tercera fase.
- Pulse Ejecutar ahora para ejecutar la microirradiatura y las imágenes de lapso de tiempo posteriores.
- Al final de la imagen de lapso de tiempo, guarde los ROI de estimulación como imágenes separadas, lo que será una ayuda útil para identificar las coordenadas de la microirradia en cualquier software posterior utilizado para el análisis.
- Tinción por inmunofluorescencia.
NOTA: El paso 5.1.3 y la Figura 2A demuestran el uso de proteínas de reparación del ADN conocidas para evaluar los tipos de lesiones de ADN introducidas por la microirradia. Ciertas lesiones de ADN también se pueden detectar mediante el uso de anticuerpos específicos después de fijar las células. También es posible detectar el reclutamiento del POI mediante la detección de anticuerpos de la proteína endógena. La visualización de γH2A.X para comprobar si hay DSB se muestra a continuación (Figura 2B). La Figura 3 muestra la consistencia de la localización y el reclutamiento de PCNA a lo largo del ciclo celular para PCNA etiquetados endógenos y exógenos.- Después del paso 5.1.3, tome solo una imagen después de la microirradiatura para garantizar un evento FRAP adecuado basado en el reclutamiento de mPlum-PCNA. Tome nota de las coordenadas exactas del FOV para encontrar el campo más tarde después del etiquetado inmunofluorescente.
- Saque la cámara de cultivo celular del microscopio e incube las células a 37 °C en una atmósfera humidificada que contenga 5% de CO2 durante 5-10 min.
NOTA: El paraformaldehído (PFA) es tóxico, y el trabajo debe realizarse en un área bien ventilada o en una campana extractora de humos. Todo el lavado e incubación posterior se realizará con volúmenes de 0,5 ml en el tobogán de la cámara de 4 pozos. Después del tiempo de incubación, lavar las células con 0,5 mL de PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4y 2 mM KH2PO4)y fijar con 0,5 mL de 4% de PFA en PBS durante 10 min a temperatura ambiente (RT). - Lave las células una vez con PBS, luego lávelas con 50 mM NH4Cl para apagar el PFA residual.
- Permeabilizar las células durante 15 min a RT con 0.1% Triton X-100 en PBS.
- Bloquee las muestras durante 1 h con tampón de bloqueo (5% FBS, 3% BSA, 0.05% Triton X-100 en PBS).
- Retire la solución de bloqueo y agregue el anticuerpo primario diluido (anti-γH2A.X, 1:2000) en el tampón de bloqueo durante 1 h en RT.
- Lavar los pozos con tampón de bloqueo 3 x 10 min.
- Añadir anticuerpo secundario diluido (anti-ratón Alexa 488 Plus conjugado, 1:2000) en el tampón de bloqueo durante 1 h en RT.
- Lavar los pozos con tampón de bloqueo 3 x 10 min.
- Contratinues el núcleo con 1 μg/ml de solución DAPI en PBS durante 15 min.
- Lave las células una vez con PBS. Las imágenes se pueden realizar directamente en PBS o en una solución de PBS con reactivos antifade (por ejemplo, AFR3) para reducir el fotobleaching.
6. Análisis de reclutamiento
NOTA: La Figura 4A muestra imágenes representativas del reclutamiento de Exo1b y PCNA en presencia de DMSO u olaparib. La Figura 4B muestra una imagen representativa para el análisis de datos. Los valores medios de fluorescencia se calcularon midiendo las intensidades medias de AcGFP utilizando un rectángulo a lo largo de la pista láser resaltado por el mPlum-PCNA (A, rectángulos amarillos) a través de diferentes puntos de tiempo utilizando Fiji. PCNA puede servir como un control interno para resaltar la irradiación exitosa a lo largo de las coordenadas ROI. Del mismo modo, también se calcularon los valores medios de fluorescencia de AcGFP para las regiones no dañadas del núcleo (B, rectángulos azules). La intensidad de la señal de fondo se midió en áreas despobladas (C, rectángulos rojos) y se restó de los valores fluorescentes medios(Figura A y B). Así, la unidad fluorescente media relativa (RFU) para cada punto de recolección de datos se calculó mediante la ecuación RFU = (A − C)/ (B − C)8,9. Los valores de RFU resultantes de la región microirradiada se normalizan a los valores de RFU antes de la microirradiación.
- Para definir la región A del sitio microirradiado, excluya de la medición las regiones nucleolares, los focos de replicación y las regiones nucleares irregulares de la célula. Mantenga pulsada la tecla Mayús entre el dibujo de dos ROI en Fiji para agrupar dos regiones separadas como una sola.
NOTA: El reclutamiento de proteínas variará entre los diferentes genes y condiciones de irradiación; por lo tanto, el tamaño de la región A debe determinarse individualmente. Una vez que se determina el ancho de píxel de la región A, debe permanecer constante para cualquier reclutamiento comparativo. En los experimentos presentados aquí, se utilizaron rectángulos de 7 píxeles de ancho. - Excluya del análisis las celdas que se movieron durante la duración de los vídeos grabados. Para incluir células altamente móviles, el análisis descrito debe llevarse a cabo fotograma a fotograma.
- Para visualizar el perfil de reclutamiento, trace los valores normalizados de RFU contra el tiempo utilizando un software estadístico.
- Calcule la diferencia en un punto de tiempo indicado entre el DMSO y el tratamiento con olaparib (n = 31) utilizando una prueba de Mann-Whitney.
Representative Results
Las células abordan cada tipo de lesión de ADN de una manera específica que también depende de la fase del ciclo celular en la que se encuentren. Por ejemplo, después de la microirradiación, las roturas de doble cadena (DSB) se procesarán mediante unión final no homóloga (NHEJ) o HR dependiendo de la fase del ciclo celular. Las nucleasas que actúan más extensamente durante las fases S y G2 del ciclo celular crean voladizos de ADN que son cruciales para la FC adecuada. Para promover la evaluación de las células en fase S, se empleó PCNA como un marcador del ciclo celular de un solo color. La Figura 1A muestra el perfil de localización de mPlum-PCNA durante la progresión del ciclo celular. El PCNA tiene una distribución completamente homogénea en el núcleo en la fase G1 y G2 (mientras que también se excluye principalmente de los nucleolos). En la fase S, el PCNA se localiza en sitios de replicación del ADN, que se pueden visualizar como puntos brillantes en el núcleo. En las células de fase S temprana, las manchas son relativamente pequeñas y están distribuidas equitativamente por todo el núcleo de la célula. Progresando a la fase S media, las manchas se vuelven borrosas y se localizan más hacia el perímetro del núcleo y los nucleolos. En la fase S tardía, las manchas se reducen en número, pero se vuelven cada vez más grandes a medida que el PCNA se concentra en los sitios de replicación tardía(Figura 1B). Es importante destacar que la expresión exógena de PCNA de la columna vertebral del vector pBABE fue menor que los niveles endógenos, pero fue suficiente para la detección por microscopía que minimiza los artefactos potenciales en la progresión del ciclo celular y DDR. La Figura 1C muestra el grado de sobreexpresión de PCNA en comparación con los niveles endógenos. Tenga en cuenta que la banda correspondiente a mPlum-PCNA migra más lentamente debido a su mayor tamaño.
Nuestro objetivo fue introducir DSB durante la microirradiatura para investigar el reclutamiento dependiente de PARP1/2 de EXO1b a estas lesiones en fase S. La Figura 2A muestra que las dosis bajas de energía (1000 μs de tiempo de permanencia) no inducen el reclutamiento de EGFP-FBXL10, un respondedor DSB (componente del complejo FRUCC 8),mientras que fue suficiente para inducir el reclutamiento de NTHL1-mCherry, una proteína de la vía de reparación por escisión de base (BER), reclutando a sitios de daño oxidativo del ADN10,11,12. A 3000 μs de tiempo de permanencia, tanto EGFP-FBXL10 como NTHL1-mCherry reclutan, demostrando una salida láser que genera lesiones oxidativas y DSB. Fortaleciendo estos resultados, la Figura 2B muestra tinción de inmunofluorescencia contra γH2A.X (marcador DSB), que es claramente más evidente cuando se usan dosis de energía más altas. PcNA sirve como un marcador del ciclo celular y un marcador para una microirradiación exitosa, ya que recluta adecuadamente con ambos ajustes de tiempo de permanencia con láser. Es importante destacar que tanto el PCNA exógeno como el endógeno marcado con proteínas fluorescentes pueden usarse para esta función de reportero, ya que se comportan de manera similar(Figura 3). El PCNA etiquetado endógenamente se diseñó insertando mRuby en el marco con el primer exón en un alelo del locusPCNA 13 (la línea celular era un regalo amable de Jörg Mansfeld).
La Figura 4A y la Figura 4C muestran el reclutamiento de EXO1b marcado con AcGFP en células de fase S. EXO1b alcanza el nivel máximo de acumulación en los sitios de micro-irradiación alrededor de 1 minuto y luego comienza lentamente a desconectarse de las lesiones de ADN después. Los enriquecimientos en sitios de microirradiación se denotan por una unidad de fluorescencia relativa > 1 en el gráfico. En presencia de olaparib, la acumulación de EXO1b en la franja láser a 1 minuto es significativamente menor en comparación con el control del vehículo. Estos resultados cons están de acuerdo con la literatura6,7. La figura 4B muestra las regiones representativas para la cuantificación (áreas A, B y C) como se describe en el punto 6 del protocolo. La Figura 4D muestra los niveles de expresión comparables de EXO1b endógeno y EXO1b-AcGFP exógeno en células utilizadas para la microirradia.
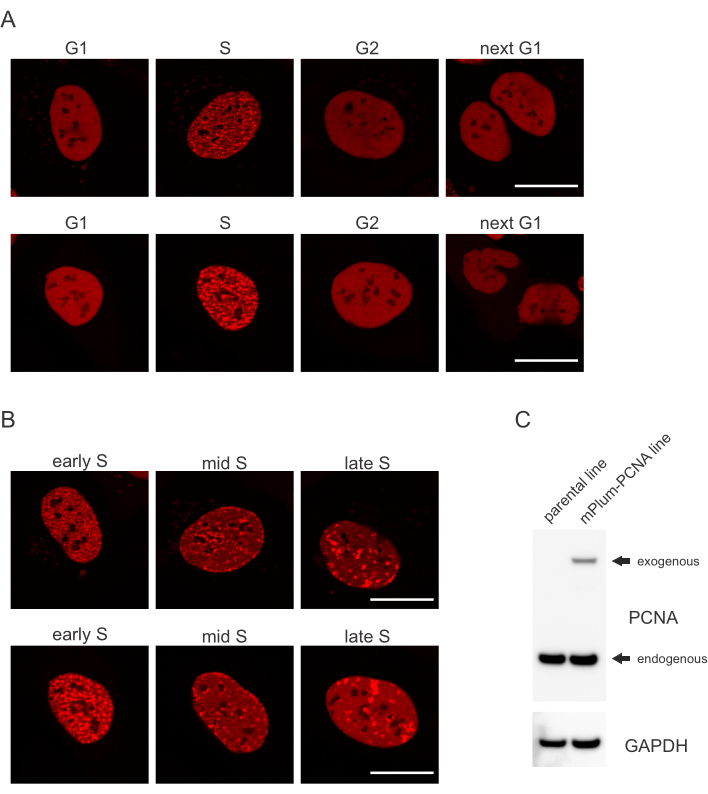
Figura 1: Patrón de localización de PCNA. (A) Las imágenes muestran el patrón de localización de PCNA exógeno e integrado de manera estable a lo largo del ciclo celular en las células del sistema operativo U-2. (B) Las imágenes muestran patrones de focos de PCNA en diferentes etapas de la fase S (temprana, media y tardía) en células de LA SO U-2. (C) Western blot que muestra niveles endógenos y exógenos de PCNA en las células del SISTEMA operativo U-2 utilizadas para la obtención de imágenes. La barra de escala representa 20 μm. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Inducción de DSB a través de la potencia de salida optimizada del láser. (A) La configuración del láser se puede optimizar para inducir diferentes formas de daño al ADN. Las células de la SO U-2 que expresan de manera estable tanto EGFP-FBXL10 como NTHL1-mCherry se utilizaron para identificar DSB y sitios de lesiones oxidativas, respectivamente. La microirradiación con una línea láser de 405 nm se llevó a cabo en células asincrónicas U-2 OS con un tiempo de permanencia de 1000 μs o 3000 μs. La barra de escala representa 20 μm. (B) La tinción inmunofluorescente contra γH2A.X se realizó en células epiteliales pigmentarias de la retina humana (hTERT RPE-1) con PCNA endógeno marcado con mRuby. Las células se fijaron y procesaron 5 minutos después de la microirradiación con un tiempo de permanencia de 1000 μs o 3000 μs. La barra de escala representa 20 μm. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Reclutamiento comparable de mRuby-PCNA endógeno y mPlum-PCNA exógeno a sitios de microirradiación a 1000 μs o 3000 μs de tiempo de permanencia del láser. Tanto el PCNA endógeno como el exógeno marcado forman focos de replicación durante la fase S. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Reclutamiento dependiente de PARP1/2 de EXO1b en fase S. Las células del sistema operativo U-2 que expresan de manera estable EXO1b-AcGFP y mPlum-PCNA fueron microirradiadas con una línea láser FRAP de 405 nm utilizando un tiempo de permanencia de 3000 μs. (A) Imágenes representativas de células microirradiadas en los puntos de tiempo indicados después del pretratamiento con control del vehículo (DMSO) u olaparib (1 μM). La barra de escala representa 20 μm. (B) Imágenes representativas de regiones definidas de áreas A, B y C para el análisis de reclutamiento. La barra de escala representa 20 μm. (C) La dinámica de reclutamiento de daños en el ADN fue capturada por imágenes de células vivas. Los valores medios relativos de fluorescencia y las imágenes se adquirieron cada 5 s durante 12 min. Para cada condición, se evaluaron ≥30 células. Los valores medios relativos de fluorescencia (líneas negras sólidas) y el error estándar (rango visualizado por un área sombreada) se trazaron contra el tiempo. La línea discontinua muestra los valores de reclutamiento a 1 min después de la microirradiatura. La diferencia entre el tratamiento con DMSO (n = 32) y olaparib (n = 31) se calculó mediante una prueba de Mann-Whitney. Astérix denota p<0.0001. (D) Western blot compara los niveles de expresión de EXO1b endógeno y EXO1b-AcGFP exógeno en células utilizadas para la microirradia. Haga clic aquí para ver una versión más grande de esta figura.
Discussion
Pasos críticos y posibles problemas/modificaciones de protocolos
El recipiente de cultivo de tejidos adecuado para la microirradiatura es fundamental para el éxito. La mayoría de los sistemas de imágenes de alta resolución están optimizados para un espesor de vidrio de cubierta de 0,17 mm. El uso de cámaras de imágenes de mayor o menor espesor o de polímeros plásticos (no optimizadas para imágenes de 405 nm) puede reducir significativamente la calidad de la imagen. Cuando use superficies de vidrio, asegúrese de que sean tratadas con cultivo de tejidos para mejorar la adhesión celular. Si no se tratan con cultivo de tejidos, estas cámaras deberán recubrirse, por ejemplo, con poli-D-lisina antes de sembrar las células. Al enchapar las células en el vidrio de cubierta con cámara, la densidad celular ideal es primordial para evitar irregularidades en el ciclo celular y estrés adicional para las células. El equilibrio térmico adecuado de los componentes del microscopio antes de la experimentación para mantener una temperatura estable es crucial tanto para mantener el enfoque a lo largo de la imagen de lapso de tiempo como para garantizar una DDR homogénea a través del tiempo y las muestras.
Es fundamental que las células estén en condiciones saludables antes de la microirradiación para reducir los datos artefactuales. Si las células tienen una morfología irregular después de la infección / selección, permita que las células progresen a través de múltiples pasajes hasta que la morfología vuelva a la normalidad. Siempre asegúrese de que las líneas de células utilizadas estén libres de contaminación por micoplasma. Entre los muchos efectos adversos de la infección por micoplasma, también causa daño en el ADN de las células huésped y podría afectar sus vías DDR14,15. La forma más sensible de detectar micoplasma en el cultivo celular es a través de PCR (versus. detección con DAPI o Hoechst).
La sobreexpresión óptima de la proteína reparadora de interés debe ser comparable a los niveles endógenos, sin embargo, lo suficientemente altos como para la detección. El promotor utilizado en los vectores virales, el título viral durante la infección y la duración del tiempo de infección se pueden ajustar para los niveles de expresión ideales. Para obtener resultados consistentes, aísle clones de células individuales para garantizar niveles de expresión homogéneos y una morfología celular normal. Se recomienda utilizar construcciones vectoriales que no sobreexpresen el PCNA etiquetado a niveles más altos que los endógenos para una función adecuada de marcadores de reparación del ciclo celular y del ADN. Incluso los niveles bajos de sobreexpresión de PCNA son suficientes para discriminar las células de fase S. Los vectores retrovirales pBABE se han utilizado con éxito para este propósito (Addgene #1764, #1765, #1766, #1767). El PCNA se puede etiquetar con cualquier proteína fluorescente roja monomérica(por ejemplo, mPlum, mCherry, mRuby, etc.) o verde monomérica (por ejemplo, mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, etc.) que luego podría combinarse con un POI etiquetado alternativamente. La sobreexpresión de un POI marcado fluorescentemente tiene algunas limitaciones y consideraciones. Las etiquetas fluorescentes pueden alterar la función normal de las proteínas y la localización. Por lo tanto, se debe considerar la ubicación de la etiqueta (N o C-terminal). Utilice siempre proteínas fluorescentes monoméricas, ya que la oligomerización de variantes no monoméricas puede afectar la función del POI.
La configuración del láser debe determinarse para cada sistema de imágenes, ya que muchos componentes de la ruta óptica afectarán la potencia real entregada a las células. La microirradiación láser puede causar varios tipos de lesiones de ADN dependiendo de la longitud de onda de excitación, la potencia de salida del láser FRAP y si se utilizó algún agente presensibilizante (como Bromodeoxiuridina o Hoechst). Los láseres de 405 nm pueden causar daño oxidativo en el ADN, roturas de cadena simple y doble16,17. Mediante el uso de configuraciones de salida láser más altas, la cantidad de DSB aumenta. En este protocolo no se utilizaron métodos de presensibilización, pero estas técnicas están muy cubiertas en la literatura y re-limitadas en la discusión a continuación. En nuestra opinión, la mejor manera de probar si se genera la lesión deseada es mediante la prueba del reclutamiento de genes específicos conocidos de la vía de daño del ADN. El reclutamiento de NTHL1 u OGG1, componentes de la vía BER, sugiere la inducción de bases de ADN oxidadas10,11,17,18,19,mientras que FBXL10 o XRCC5 indican la presencia de DSB8,20,21. El reclutamiento de XRCC1 puede indicar tanto la presencia de bases de ADN oxidadas como roturas monocatenadas (SSB)22,23. XPC (es decir, RAD4) es un buen indicador de NER que elimina los voluminosos aductos de ADN generados por la luz ultravioleta (UV)17,24. Debido a que el reclutamiento de proteínas exógenas puede introducir ciertas irregularidades, la tinción inmunofluorescente de proteínas o marcadores endógenos de reparación del ADN (como γH2A.X para roturas de doble cadena) puede confirmar la presencia de lesiones específicas de ADN. Alternativamente, también se podrían usar anticuerpos criados contra tipos específicos de lesiones de ADN. Para ajustar la potencia láser entregada, se puede cambiar tanto el tiempo de permanencia como la potencia del láser.
Con la ayuda de modelos matemáticos, se podría realizar un análisis cinético detallado que puede proporcionar información valiosa sobre las propiedades de reclutamiento del POI (por ejemplo, la contribución de múltiples dominios de unión al ADN, la sensibilidad hacia diferentes eventos de señalización, etc.). La evaluación automatizada del reclutamiento y el seguimiento celular podrían combinarse para crear flujos de trabajo robustos 1,25.
Ventajas y limitaciones de la presensibilización del ADN
La presensibilización del ADN antes de la microirradiación es una herramienta comúnmente utilizada para el reclutamiento de proteínas de reparación del ADN16,17. Sensibilizar el ADN antes de la microirradiación lo deja más susceptible a los DSB. Los dos métodos más comunes para la presensibilización del ADN son el pretratamiento de las células con bromodeoxiuridina (BrdU) o colorante Hoechst. Para los sistemas que no son capaces de microirradiarse a altas potencias láser, estos métodos pueden ser necesarios para inducir lesiones de ADN como los DSB. Además, en ausencia de un detector de luz transmitida o una señal fluorescente que resalte el núcleo celular (por ejemplo, cuando se estudia el reclutamiento de proteínas endógenas de reparación del ADN sin etiquetar), Hoechst actúa como una herramienta de presensibilización y una tinción nuclear fluorescente. Sin embargo, la presensibilización del ADN puede introducir complicaciones significativas. BrdU (utilizado a una concentración final de 10 μM) debe agregarse a las células 24 horas (o tiempo equivalente a un ciclo celular completo en la línea celular utilizada) para incorporarse adecuadamente en el ADN y puede causar interferencias en el ciclo celular26. Hoechst 33342 (utilizado a una concentración final de 1 μg/ml) es citotóxico después de largos períodos de incubación, pero requiere tiempo suficiente para saturar el núcleo con el colorante. Por lo tanto, solo debe aplicarse 15-20 minutos antes de la microirradia; de lo contrario, los datos de contratación no serán coherentes. Las células teñidas de esta manera no se pueden mantener en cultivo durante más de unas pocas horas27,28. Asegúrese de no usar Hoechst 33358, que no es tan permeable a las células como el tinte Hoechst 33342. La presensibilización también puede introducir una variación innecesaria entre los experimentos y hace que el experimento sea aún más sensible a las diferencias en la densidad celular (ya que esto afectará la cantidad de tinte / célula incorporado).
Ventajas y limitaciones de la microscopía confocal
La velocidad de obtención de imágenes de la microscopía confocal puede ser limitante en comparación con la microcopia de campo amplio. Sin embargo, un microscopio confocal equipado con un escáner resonante puede mejorar enormemente la velocidad de imagen (a costa de la resolución) acercándose a las velocidades de la microscopía de disco giratorio. Tres características hacen que el sistema confocal A1R HD25 sea una excelente opción para el protocolo presentado aquí. En primer lugar, el FOV de 25 mm del sistema permite obtener imágenes de entre 15-20 celdas en un solo campo escaneado (frente a 5-10 celdas en configuraciones regulares), lo que limita el número de adquisiciones necesarias para obtener suficientes células para el análisis estadístico. En segundo lugar, el módulo FRAP y dos cabezales de escaneo permiten obtener imágenes y microirradiar las células simultáneamente, no solo secuencialmente. Por último, la flexibilidad de tener los escáneres resonantes y galvanos proporciona la capacidad de cambiar fácilmente entre imágenes de alta resolución temporal con una velocidad excepcional que minimiza el enfriamiento de los fluoróforos, e imágenes de alta resolución espacial que utilizan velocidades de escaneo más lentas para producir imágenes con una mayor relación señal/ruido. Si bien el sistema utilizado permitió la flexibilidad antes mencionada, para parecerse a configuraciones de microscopio confocal más ampliamente disponibles, solo se utilizó el escáner galvano en los experimentos presentados (tanto para microirradiación como para imágenes posteriores).
Ventajas y limitaciones de la microirradiatura
Si bien la microirradiación proporciona una resolución espacial y temporal sin igual, no está exenta de limitaciones. El daño al ADN por microirradiación láser está altamente agrupado en partes específicas del núcleo en comparación con los agentes dañinos naturales. Por lo tanto, la respuesta de la cromatina debido a la microirradiación puede diferir en comparación con el daño distribuido homogéneamente. Además, la microirradiación consume mucho tiempo y solo puede llevarse a cabo en unas pocas docenas de células, mientras que los grandes métodos bioquímicos basados en la población (fraccionamiento de cromatina, inmunoprecipitación, ChIP) pueden proporcionar una mayor robustez al estudiar miles de células a la vez. La verificación de las observaciones realizadas por microirradiación con técnicas bioquímicas tradicionales es una estrategia eficaz para obtener conclusiones fiables. Aunque es posible la microirradia simultánea de muchas células en un determinado FOV, el sistema de imágenes necesitará más tiempo para realizar la tarea. Por lo tanto, la medición de la dinámica de las proteínas que reclutan muy rápidamente a las lesiones de ADN limita el número de posibles ROI para la microirradia utilizada simultáneamente. En el sistema de imágenes utilizado para este protocolo, la microirradia de un solo ROI de 1024 píxeles de largo toma 1032 ms usando un tiempo de permanencia de 1000 μs y 3088 ms usando un tiempo de permanencia de 3000 μs para completarse. El uso de múltiples líneas de ROI aumentará significativamente el tiempo necesario para finalizar la microirradiatura (por ejemplo, el ROI de 7 x 1024 píxeles de largo toma 14402 ms usando un tiempo de permanencia de 1000 μs y 21598 ms usando un tiempo de permanencia de 3000 μs). Este tiempo se pierde por la adquisición de imágenes y debe tenerse en cuenta. Al obtener imágenes de eventos de reclutamiento rápido, use el ROI más corto posible y solo microirradie una célula a la vez.
Ventajas y limitaciones sobre los métodos de sincronización
Para los estudios específicos del ciclo celular, los métodos existentes implican la sincronización de las células en fases específicas del ciclo celular o el uso de reporteros fluorescentes para identificar la fase específica del ciclo celular de la célula. Sin embargo, cada uno de estos métodos proporciona sus propios desafíos y limitaciones.
El sistema FUCCI3 (que se basa en formas truncadas marcadas con proteínas fluorescentes de CDT1 y Geminin) es una herramienta particularmente útil para estudios del ciclo celular, pero tiene limitaciones cuando se trata de diferenciar entre las fases S y G2 del ciclo celular. Los niveles de Geminina ya son altos desde mediados de la fase S y se mantienen altos hasta la fase M, lo que hace que estas fases sean difíciles de separar. El uso del sistema FUCCI también significa que no se pueden utilizar dos canales ópticos del microscopio para obtener imágenes del POI.
Las líneas celulares no cancerosas podrían sincronizarse en G0 mediante la eliminación de los factores de crecimiento que se encuentran en el suero (inanición sérica) que causan poco o ningún daño en el ADN de las células. Sin embargo, la mayoría de las líneas celulares de cáncer continuarán progresando parcialmente a través del ciclo celular, incluso sin cantidades adecuadas de suero en sus medios. Además, las células comienzan a perder parcialmente la sincronización a finales de G1, fase S temprana. Además de la inanición sérica, existen numerosos métodos químicos para lograr la sincronización del ciclo celular. La hidroxiurea, la puldicolina y los bloques de timidina son métodos para detener la replicación del ADN para sincronizar las células en la fase S temprana. Si bien estos métodos son baratos y simples, introducen estrés de replicación que resulta en daño en el ADN. Se ha demostrado que estos inhibidores de la replicación del ADN inducen la fosforilación de H2A. X, un marcador bien conocido de losOSD 2,29. El método de usar PCNA etiquetado como marcador para las células de fase S reduce el potencial de artefactos causados por la sincronización química y se puede aplicar a una amplia gama de líneas celulares en comparación con la inanición sérica.
Conclusión
El daño al ADN es una fuerza impulsora para las enfermedades genéticas donde las lesiones mutagénicas pueden conducir a la transformación maligna de las células. Dirigirse a la maquinaria de síntesis de ADN es una estrategia terapéutica fundamental en el tratamiento de enfermedades hiperproliferativas como el cáncer. Para tratar estas enfermedades de una manera más específica, necesitamos una mejor comprensión de las proteínas que reparan las lesiones de ADN. El protocolo descrito aquí ayuda a los estudios basados en microirradiación en fase S al minimizar los desafíos presentados por los métodos de sincronización tradicionales para reducir los posibles artefactos y aumentar la reproducibilidad de los experimentos.
Disclosures
Los autores afirman que la publicación del trabajo presentado fue patrocinada por Nikon Corporation. Los autores declaran que no existen intereses contrapuestos.
Acknowledgments
Los autores agradecen a M. Pagano por su continuo apoyo, así como a D. Simoneschi, A. Marzio y G. Tang por su revisión crítica del manuscrito. B. Miwatani-Minter agradece a R. Miwatani y B. Minter por su continuo apoyo. G. Rona agradece a K. Ronane Jurasz y G. Rona por su continuo apoyo.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
