Previously, we successfully established a MSPP scheme for C. jejuni ssp. jejuni13. Here, we aimed to extend the method to the sibling subspecies C. jejuni ssp. doylei. In this specific setting, seven C. jejuni ssp. doylei isolates were acquired from the Belgian collection of microorganisms/Laboratory of Microbiology UGent BCCM/LMG Ghent, Belgium. All seven isolates used for our analyses were of human origin. The genome-sequenced strain ATCC 49349 (LMG 8843), originally isolated from feces of a 2-year-old Australian child with diarrhea served as the reference. From the collection, six additional strains were available: LMG 9143, LMG 9243, and LMG 9255 isolated from feces of children suffering diarrhea and living in Brussels, Belgium; LMG 7790 (ATCC 49350) isolated from a gastric biopsy sample of an individual from Germany; and LMG 8870 (NCTC A613/87) as well as LMG 8871 (NCTC A603/87), both isolated from different blood samples drawn from South African children.
Todos C. jejuni ssp. doylei isolates were stored at -80 °C as cryobank stocks, freshly thawed for each analysis and cultured by using Columbia agar base supplemented with 5% sheep blood and incubation for 48 hr at 37 °C under microaerophilic conditions (5% O2, 10% CO2, 85% N2).
In contrast to the establishment of the MSPP technique for C. jejuni ssp. jejuni13 it was not possible to base the method on an isoform database derived from a larger sequence collection. Only a single C. jejuni ssp. doylei entry was present in the rMLST database, and this corresponded to the reference strain C. jejuni ssp. doylei ATCC 4934927. Therefore, each biomarker mass shift detected in comparison to strain C. jejuni ssp. doylei ATCC 49349 was a new entry in the isoform database and needed to be reconfirmed by Sanger sequencing.
To analyze the diversity of the obtained C. jejuni ssp. doylei isolates, MLST was performed and a MLST-based UPGMA-dendrogram including the seven examined C. jejuni ssp. doylei isolates and C. jejuni ssp. jejuni strain 81-176 as the outgroup calculated (Figure 1). Each C. jejuni ssp. doylei isolate belonged to a different MLST-sequence type, falling into two subclusters. Strain LMG7790 had the longest phylogenetic distance as compared to the remaining C. jejuni ssp. doylei isolates.
The recordable MALDI-TOF reference mass spectrum of C. jejuni ssp. doylei ATCC 49349 contained 14 singularly charged biomarker ions that could be identified by comparison of the calculated molecular masses with the reference spectrum (Figure 2).
Within the collection, varying isoforms were detected for L32-M, L33, the hypothetical protein encoded by gi|152939117, S20-M, and S15-M (Figure 3). The remaining masses assigned to biomarker ions were invariable in the tested C. jejuni ssp. doylei isolates. The amino acid substitution corresponding to the mass shifts have been identified by PCR-amplification and Sanger sequencing of the particular gene using the primers listed in Table 3. Table 4 lists the amino acid sequences of all biomarker ions included in the MSPP scheme as well as the detected allelic isoforms.
A MSPP-based phyloproteomic UPGMA-tree (Figure 4) was calculated for the same seven C. jejuni ssp. doylei isolates and C. jejuni ssp. jejuni strain 81-176 as the outgroup using the concatenated amino acid sequences of all 14 biomarker ions in order of their molecular weight in the reference strain. Each isolate represented an individual MSPP-sequence type. Although the obtained phyloproteomic relations were not fully identical to the ones obtained by MLST, the C. jejuni ssp. doylei isolates again arranged in two subclusters. The first subcluster was formed by LMG 8843, LMG 8870, and LMG 9243, the second by LMG 9255, LMG 9143, LMG 8871 and LMG 7790. As seen with the MLST-analysis, isolate LMG7790 showed the longest phyloproteomic distance in the MSPP-analysis.
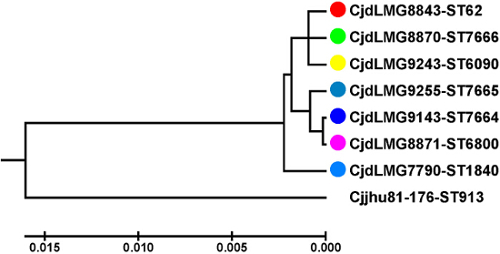
Figure 1: MLST-based phylogenetic UPGMA-tree. Balanced MLST-based UPGMA-dendrogram constructed from seven C. jejuni ssp. doylei isolates and C. jejuni ssp. jejuni strain 81-176. Strain color code: LMG 8843 (black), LMG 8870 (grey), LMG 9243 (blue), LMG 9255 (turquoise), LMG 9143 (green), and LMG 8871 (pink), LMG 7790 (yellow), and 81-176 (white). The MLST-sequence type is given behind the name of each isolate. Every C. jejuni ssp. doylei isolate belongs to a different MLST-sequence type. X-axis indicates the linkage distances. Strain LMG 7790 shows the longest phylogenetic distance compared to the remaining six isolates. Strains LMG 8843, LMG 8870, and LMG 9243 as well as LMG 9255, LMG 9143, and LMG 8871 form two different subclusters within the C. jejuni ssp. doylei cluster. Please click here to view a larger version of this figure.
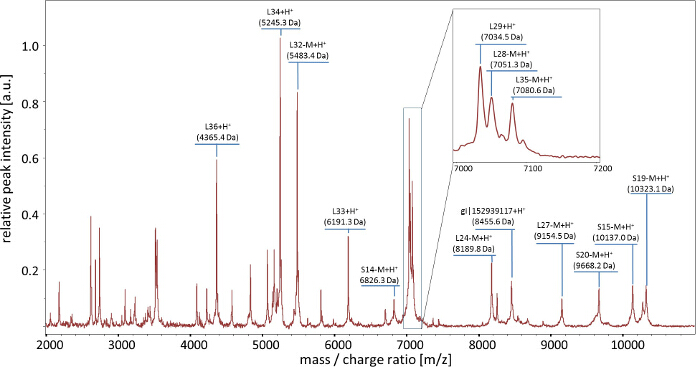
Figure 2: Tentative assignment of genomic correlates of C. jejuni ssp. doylei ATCC 49349 (LMG 8843) to observed biomarker masses. Based on the calculated masses (average isotopic composition) of predicted ORFs from the whole genome sequence of C. jejuni ssp. doylei strain ATCC 49349, biomarker masses were assigned to the corresponding protein coding sequences. However, within measurement range, the biomarker masses for the ribosomal subunits L31 (MW = 7,315 Da), S17 (MW = 9,549 Da), and S18 (MW = 10,285 Da) as well as their de-methioninated isoforms (inset m/z = 7,000-7,200 Da) could not be unambiguously assigned. Therefore, L31, S17, and S18 were not included in the C. jejuni ssp. doylei MSPP scheme. Please click here to view a larger version of this figure.
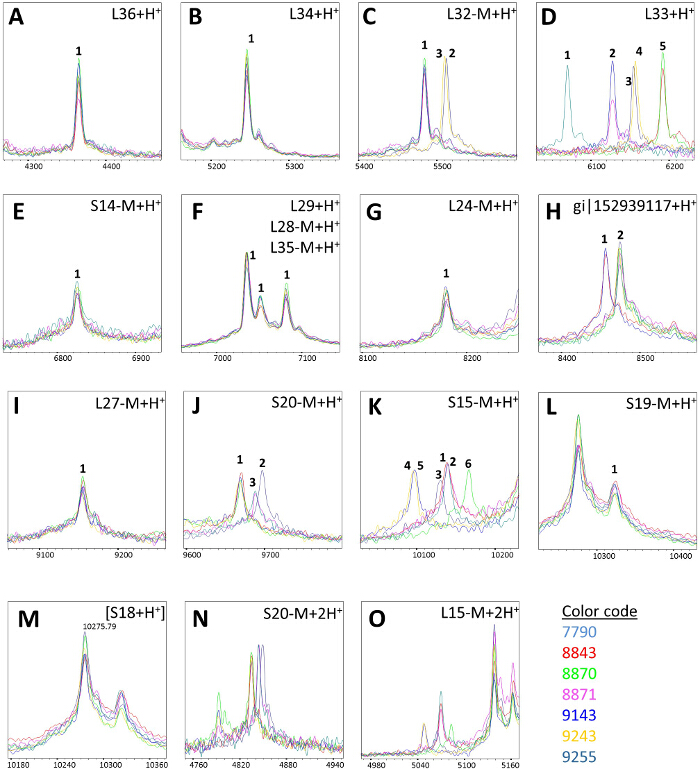
Figure 3: Specific biomarker mass peaks of the C. jejuni ssp. doylei MSPP scheme. In each panel the mass spectra of all seven tested C. jejuni ssp. doylei isolates (color code as in Figure 1) corresponding to the particular MSPP types have been overlaid to indicate biomarker mass shifts due to allelic isoforms: (A) L36+H+; (B) L34+H+; (C) L32-M+H+; (D) L33+H+; (E) S14-M+H+; (F) L29+H+, L28-M+H+, and L35-M+H+; (G) L24-M+H+; (H) gi|152939117+H+; (I) L27-M+H+; (J) S20-M+H+; (K) S15-M+H+; (L) S19-M+H+; (M) an intense mass obscuring variable potential biomarker peak of ribosomal protein S18; (N) S20-M+2H+; (O) L15-M+2H+; X-Axes: mass [Da]·charge-1 ratio, scale 200 Da. Y-Axes: intensity [arbitrary units], spectra were individually adjusted to a comparable noise level for better visualization of low-intensity peaks. "-M" after the name of a ribosomal subunit indicates the de-methioninated isoform. Please click here to view a larger version of this figure.
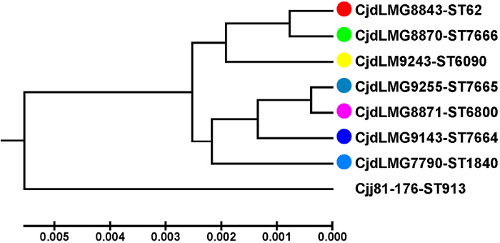
Figure 4: MSPP-based phyloproteomic UPGMA-tree. The depicted phyloproteomic UPGMA-tree includes the same seven C. jejuni ssp. doylei isolates and C. jejuni ssp. jejuni strain 81-176 as in Figure 1. A colored spot (color code as in Figure 1) indicates each strain. For better comparison the isolates are arranged in input order, which was the order obtained from the MLST-based tree. The axis below the dendrogram indicates the linkage distances. Although the obtained phyloproteomic relations are not completely identical to the phylogenetic MLST-based tree, the global picture is comparable. The population splits into two groups: the three isolates LMG 8843, LMG 8870, and LMG 9243 form one cluster, while LMG 9255, LMG 9143, LMG 8871 and LMG 7790 form a second cluster. Within this cluster LMG 7790 shows the longest phyloproteomic distance compared to the subcluster formed by LMG 9255, LMG 9143, and LMG 8871. Within this subcluster LMG 9143 switches position in relation to LMG 9255. However, even using the MSPP-method each isolate represents an individual MSSP-sequence type. Please click here to view a larger version of this figure.
Table 1: Calculated masses of all annotated ORFs of the C. jejuni ssp. doylei ATCC 49349 strain. List of all calculated monoisotopic molecular weights of each protein encoded in the C. jejuni ssp. doylei ATCC 49349 genome. The ExPASy Bioinformatics Resource Portal was used for calculation of the particular molecular masses. Column B lists the methioninated isoforms whereas column C lists the de-methioninated isoforms. Biomarker masses included in the C. jejuni ssp. doylei MSPP scheme are highlighted in red. Note: The biomarker protein L36 is not annotated in the genome sequence of C. jejuni ssp. doylei ATCC49349. Please click here to download this file.
Table 2: Mass changes induced by amino acid changes due to single SNPs. All potential single nucleotide polymorphisms were checked for the resulting amino acid exchange using the standard genetic code. All silent mutations were discarded, and nonsynonymous mutations compiled together with the resulting mass change. Please click here to download this file.
Table 3: Oligonucleotide primers used for sequencing of the C. jejuni ssp. doylei genes included in the MSPP-scheme Please click here to download this file.
Table 4: Overview of all isoforms included in the C. jejuni ssp. doylei MSPP-scheme. This table lists all biomarker ions included in the C. jejuni ssp. doylei MSPP scheme. The amino acid sequence of the reference strain ATCC 49349 is given completely. For further detectable isoforms, only specific amino acid substitutions are listed. The amino acid numbering always includes the start-methionine; if mass spectrometry indicates its absence, it is written in brackets (M). For each isoform molecular mass, mass difference to reference strain ATCC 49349 isoforms and frequency within the isoform dataset is indicated. Please click here to download this file.
