The immuno-RNA-FISH protocol described in this manuscript was carried out using two cellular systems: a Vero cell line and a 3D HAE culture. The major steps for both cellular models are shown in Table 2. The RNA-FISH protocol for visualization of SARS-CoV-2 in HAE cultures includes steps that are typical for tissue samples, i.e., permeabilization with 100% MeOH and rehydration through a graded series of MeOH-PBS and 0.1% Tween solutions. Immunofluorescence was performed after RNA-FISH was complete. Zstack images were acquired and processed.
Figure 1 shows immuno RNA FISH in mock-inoculated control Vero cells or cells infected with SARS-CoV-2. Figure 2 shows immuno RNA FISH in mock-inoculated control HAE cultures or cultures infected with SARS-CoV-2. Figure 3 shows optimization of the permeabilization protocol in Vero cells: 70% ethanol overnight at -20 °C or 0.1% Tween-20 in PBS for 5 min at RT. Permeabilization with detergent results in a clear, specific signal for SARS-CoV-2 subgenomic RNA, whereas using ethanol results in a blurry unfocused image.
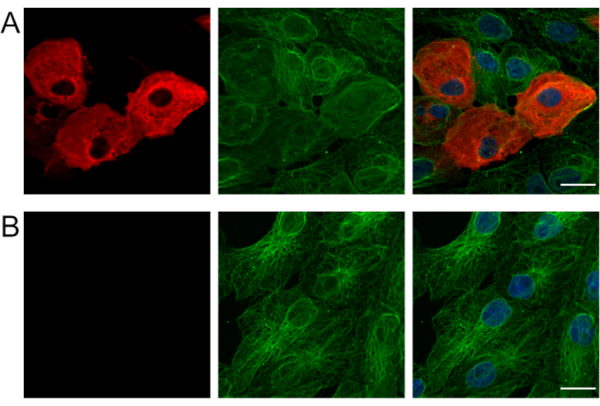
Figure 1: Immuno-RNA-FISH to detect SARS-CoV-2 RNA and β-tubulin in Vero cells. Localization of SARS-CoV-2 subgenomic RNA in (A) infected and (B) mock-inoculated Vero cells. Viral RNA was visualized by FISH (red). β-tubulin is stained with antibodies against mouse β5tubulin (1:100, overnight incubation at 4 °C) and with Alexa fluorophore 488-conjugated secondary antibodies (1:400, 1 h incubation at RT).Nuclei were stained with DAPI (blue). Each image is a single confocal plane. Scale bar = 20 µm. Abbreviations: SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; FISH = fluorescence in situ hybridization; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
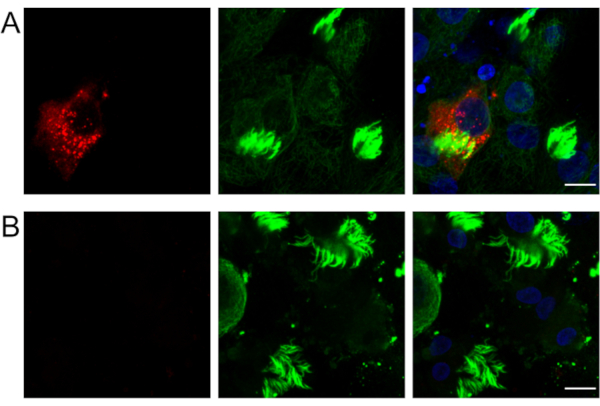
Figure 2: Human airway epithelial cells infected with SARS-CoV-2. Localization of SARS-CoV-2 subgenomic RNA in (A) infected and (B) mock-inoculated HAE cultures. Viral RNA was visualized by FISH (red). Ciliated cells are visualized using antibodies against mouse β5-tubulin (1:100, overnight incubation at 4 °C) and with Alexa fluorophore 488-conjugated secondary antibodies (1:400, 1 h incubation at RT). Nuclei were stained with DAPI (blue). Each image represents a max projection reconstructed from confocal image stacks (thickness = 3 µm). Scale bar = 10 µm. Abbreviations: SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; FISH = fluorescence in situ hybridization; HAE = human airway epithelium; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
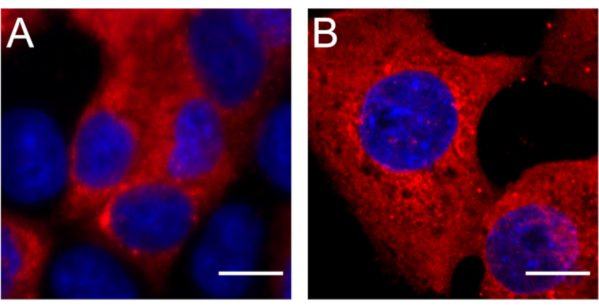
Figure 3: Optimization of permeabilization conditions for Vero cells. Permeabilization of Vero cells with (A) 70% ethanol and (B) with 0.1% Tween-20 in PBS. Permeabilization with detergent results in a clear specific signal for SARS-CoV-2 subgenomic RNA, whereas ethanol results in a blurry image. Viral RNA is shown in red. Nuclei were stained with DAPI (blue). Each image represents a max projection reconstructed from confocal image stacks (thickness = 3 µm). Scale bar = 10 µm. Abbreviations: SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2; PBS = phosphate-buffered saline; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
Supplemental Figure 1: SARS-CoV-2 N gene sequence (5'-3') Please click here to download this file.
| Buffer | Volume of methanol [mL] | Volume of PBST [mL] |
| 75% MeOH/25% PBST | 75 | 25 |
| 50% MeOH/50% PBST | 50 | 50 |
| 25% MeOH/75% PBST | 25 | 75 |
| 100% PBST | 0 | 100 |
| Total | 100 mL | |
Table 1: Preparation of gradient methanol/PBST solutions for rehydration. To rehydrate human airway epithelium samples after overnight incubation in absolute methanol (MeOH), a slow exchange of the environment is necessary. To do this, slow exchange must occur by incubating with buffers in which the proportions of MeOH and PBST (0.1% Tween-20 in 1x phosphate-buffered saline) change gradually. Reagent volumes sufficient to prepare 100 mL of each solution, enough to perform several experiments, are listed.
| Module | Step | Vero cells | HAE cultures |
| RNA Fluorescence in situ hybridization (RNA FISH) | Fixation | (3.7% PFA) 10-40 min at room temperature or overnight at room temperature | |
| Permeabilization | (PBST: 0.1% Tween-20 in 1x PBS) 10 min at room temperature | (0.1% Tween-20 in 1x PBS) 2 × 5 min at room temperature | |
| (100% MeOH) overnight at -20 °C | |||
| Rehydration | (Graded methanol (MeOH)/PBST) 5 x 5 min, 50% 5x SSCT/PBST wash 5 min, 5x SSCT wash 5 min on ice | ||
| Detection (pre-hybridization) | (Probe hybridization buffer) 30 min at 37 °C, 200-300 µL | (Probe hybridization buffer) 5 min on ice, then 30 min at 37 °C, 100 µL | |
| Detection | (Probe solution) 12-18 h at 37 °C, 30 – 50 µL | (Probe solution) 12-18 h at 37 °C, 100 µL | |
| Probe washings | (Probe wash buffer) 4 x 5 min | (Probe wash buffer) 4 x 15 min | |
| (5 × SSCT) 2 x 5 min | |||
| Amplification (pre-amplification) | (Amplification buffer) 30 min at room temperature, 200-300 µL | (Amplification buffer) 30 min at room temperature, 100 µL | |
| Amplification | (Amplifiers solution) 12-18 h at room temperature in dark place, 30-50 µL | (Amplifiers solution) 12-18 h at room temperature in dark place, 100 µL | |
| Amplifiers washing | (5x SSCT) 5 x 5 min | (5x SSCT) 2 x 5 min, 2 x 15 min, 1 x 5 min | |
| ImmunoFluorescence (IF) | Blocking | (1% BSA in PBST) 30 min at 37 °C | |
| Primary antibody incubation | (Antibody solution of apropriate concentration in blocking solution) 2 h at room temperature / overnight at 4 °C, 30-50 µL | (Antibody solution of apropriate concentration in blocking solution) 2 h at room temperature / overnight at 4 °C, 100 µL | |
| Primary antibody washing | (PBST) 3 x 5 min at room temperature | ||
| Secondary antibody incubation | (Antibody solution of apropriate concentration in blocking solution) 1 h at 37 °C, 30-50 µL | (Antibody solution of appropriate concentration in blocking solution) 1 h at 37 °C, 100 µL | |
| Secondary antibody washing | (PBST) 3 x 5 min at room temperature | ||
| Nuclear staining | (DAPI solution) 10 min at room temperature, then 2 x 5 min with 1x PBS | ||
Table 2: Workflow of the Immuno-RNA-FISH protocol in cell lines and HAE cultures. Immuno-RNA-FISH is feasible in both cellular models, but requires different approaches. The main steps are shown, along with the buffers used (in parentheses), followed by the duration and temperature of incubation. In several steps, critical differences in the volume of incubation reagent per sample are given to simplify the calculations. If the volume is not specified, it is selected arbitrarily so that it completely covers the sample (usually 200 µL) with agitation. Abbreviations: FISH = fluorescence in situ hybridization; HAE = human airway epithelium; PFA = paraformaldehyde; DAPI = 4′,6-diamidino-2-phenylindole; BSA = bovine serum albumin; PBS = phosphate-buffered saline; MeOH = methanol.
