We optimized the protocol for me1Ψ-UTP modified mRNA production, liposome preparation, and mRNA transfection experiments with cationic liposomes into multiple mammalian cells (Figure 1). To synthesize mRNA, the mammalian codon-optimized eGFP IVT template was amplified from the mEGFP-N1 mammalian expression vector and purified by organic extraction/ethanol precipitation method (Figure 2). Later, me1Ψ-UTP modified RNA and mRNA were produced by the IVT process. The denaturing RNA agarose gel electrophoresis data showed that these synthesized RNAs had good integrity and the correct length (750 base RNA, and ~1000 base mRNA with respect to the RNA ladder) (Figure 3).
To prepare cationic liposomes, a thin-film hydration method and sonication were used to form small unilamellar vesicles (SUVs). Physico-chemical characterization of the liposomes revealed that the hydrodynamic diameters were observed around 65 nm and the surface potentials were around +20 meV (Figure 4). A gel retardation assay was performed with liposome-mRNA complexes at varying charge ratios of lipid/base from 1:1 to 8:1. The cationic liposomes showed a high binding efficiency to mRNAs even at a 1:1 charge ratio (Figure 5). Hence, we used a 1:1 charge ratio for mRNA transfection experiments. Liposome mediated eGFP mRNA transfection experiments were performed in HEK-293T and NIH/3T3 cell lines. eGFP expression was analyzed with flow cytometry. me1Ψ-UTP modified mRNA showed superior and stable eGFP protein expression when compared to unmodified mRNA in HEK-293T and NIH/3T3 cells on the 3rd-day post-transfection (Figure 6, 7).
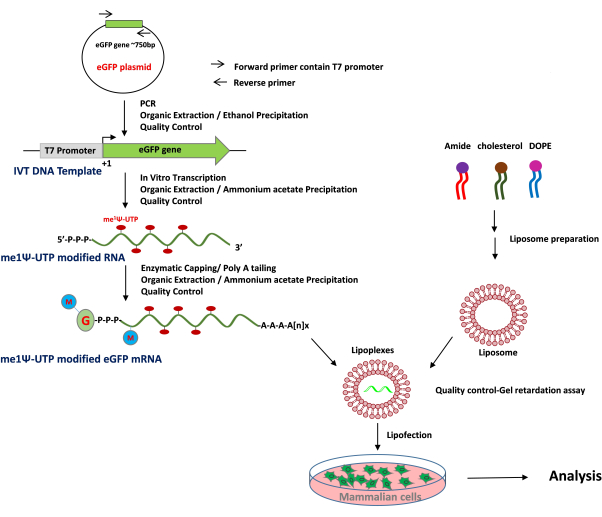
Figure 1: Schematic presentation of me1Ψ-UTP modified mRNA production, liposome preparation and transfection protocol. The IVT DNA Template (T7 promoter-Gene ORF) is amplified by PCR and purified. me1Ψ-UTP modified mRNAs are generated by the IVT process using the IVT DNA template and purified. The cationic liposome is prepared and complexed with me1Ψ-UTP modified mRNA (Lipoplex) and can be transfected into mammalian cells. Please click here to view a larger version of this figure.
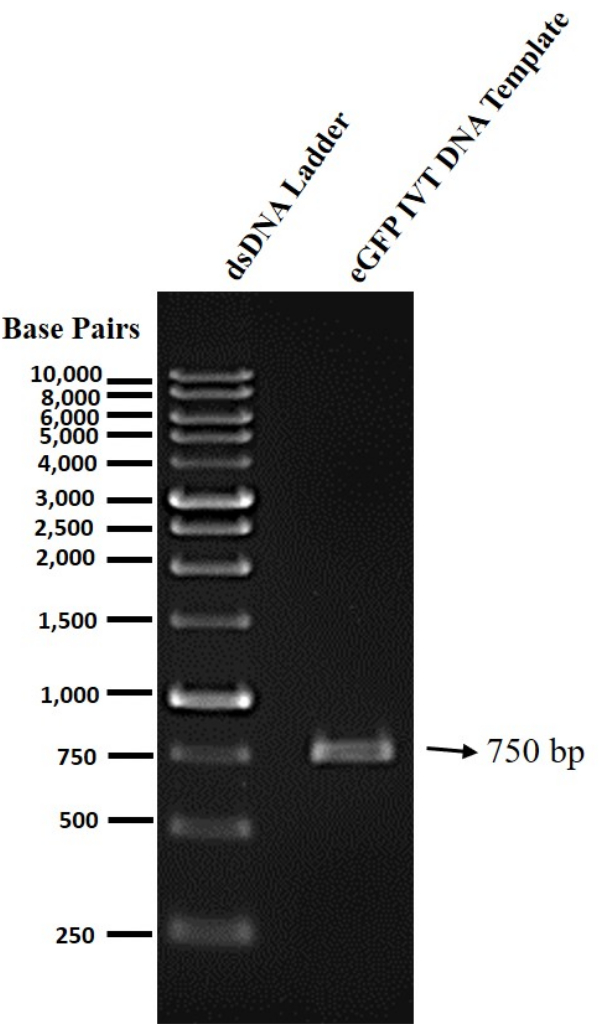
Figure 2: Determination of IVT DNA template quality in agarose gel electrophoresis. The purified eGFP IVT DNA template was run on a 1% agarose gel and visualized by gel. Please click here to view a larger version of this figure.
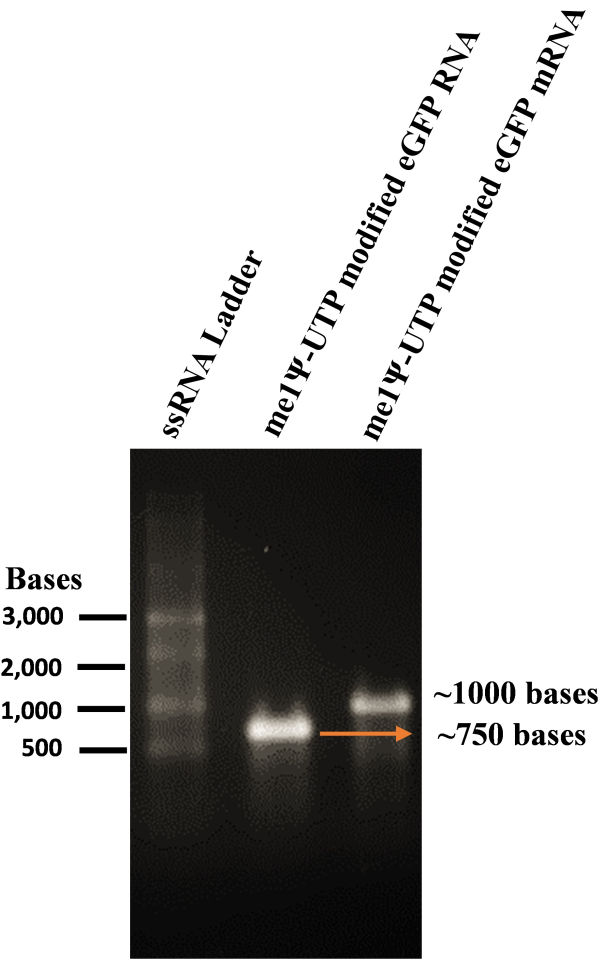
Figure 3: Analysis of size and quality of me1Ψ-UTP modified IVT RNA by denaturing RNA electrophoresis in agarose gel. The purified me1Ψ-UTP modified RNAs were denatured and loaded on a 1% TAE-agarose gel, and the size and quality of RNAs were determined by gel. Lane 1: ssRNA ladder, Lane 2: me1Ψ-UTP modified IVT RNA, Lane 3: me1Ψ-UTP modified IVT RNA with Cap1 and Poly A tail.Please click here to view a larger version of this figure.
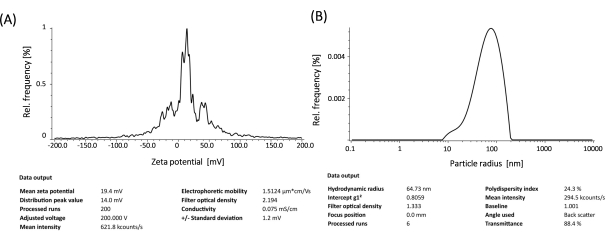
Figure 4: Physico-chemical characterization of the liposomes: Surface potentials (A) and hydrodynamic diameters (B). Please click here to view a larger version of this figure.
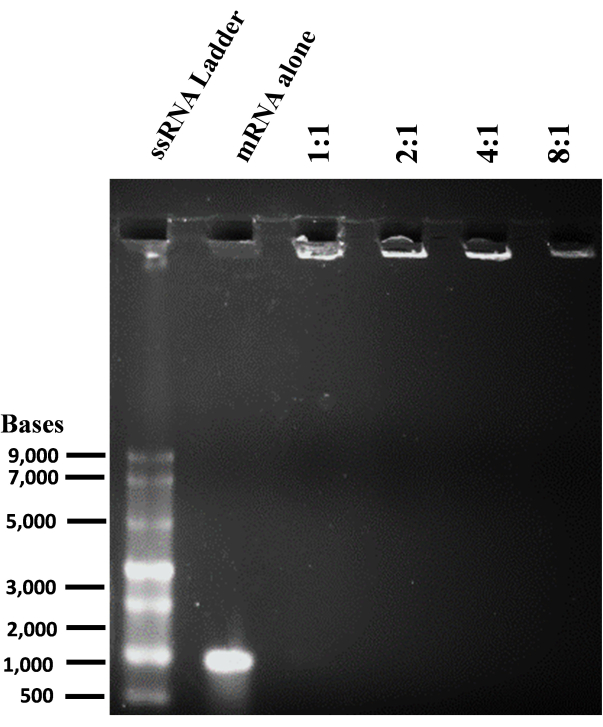
Figure 5: mRNA binding ability of liposome was determined by denaturing agarose gel retardation assay. Liposome-mRNA complexes (Lipoplexes) were prepared at different lipid/base charge ratios and loaded on a 1% agarose gel and gel documented. Please click here to view a larger version of this figure.
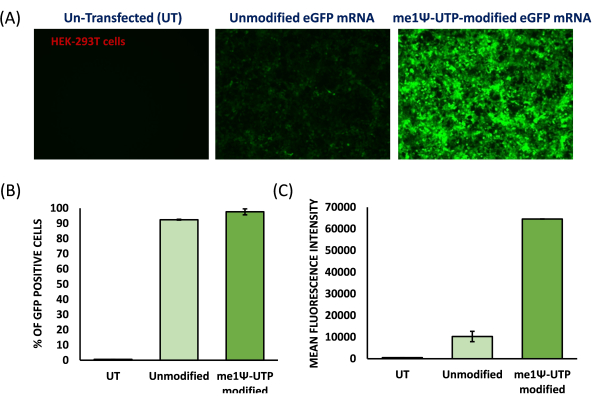
Figure 6: Protein expression efficiency of me1Ψ-UTP modified eGFP mRNA in human HEK-293T cells. (A) Fluorescence images of unmodified eGFP mRNA and me1Ψ-UTP-modified eGFP mRNA transfected with cationic liposomes into HEK-293T cells obtained on 3rd-day post-transfection (100x Magnification). % of eGFP protein expression (B) and mean fluorescent intensity (MFI). (C) of the transfected cellswere analyzed using flow cytometry. (N=3) Please click here to view a larger version of this figure.
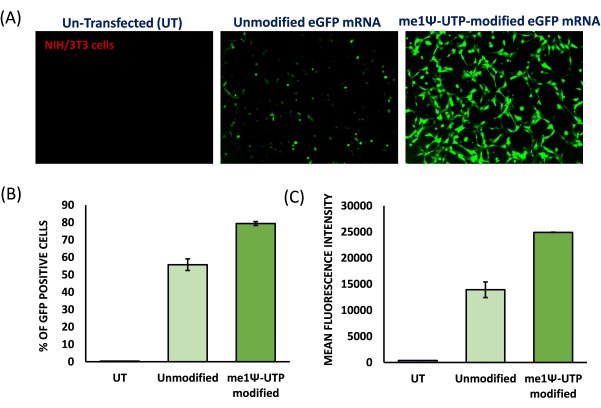
Figure 7: Translation efficiency of me1Ψ-UTP modified eGFP mRNA in mouse NIH/3T3 cells. (A) Fluorescence images of unmodified eGFP mRNA and me1Ψ-UTP-modified eGFP mRNA transfected into NIH/3T3 cells obtained on the 3rd-day post-transfection (100x Magnification). % of eGFP protein expression (B) and mean fluorescent intensity (MFI). (C) of the transfected cellswere analyzed using flow cytometry. (N=3) Please click here to view a larger version of this figure.
| Components | 25 µL reaction | Final concentration |
| 5x Q5 buffer | 5 µL | 1x |
| 10 mM dNTP | 0.5 µL | 200 µM |
| 10 µM Forward primer | 1.25 µL | 0.5 µM |
| 10 µM Reverse primer | 1.25 µL | 0.5 µM |
| Q5 polymerase | 0.25 µL | 0.02 U/µL |
| Gene of interest in plasmid (template) | 1-5 ng | variable |
| Nuclease free water | To 25 µL |
Table 1: PCR reaction mixture preparation
| Steps | Duration | Temperature | Cycle number |
| Initial denaturation | 30 seconds | 98 °C | 1 |
| Denaturation | 10 seconds | 98 °C | |
| Annealing | 20 seconds | variable | 18-25 |
| Extension | variable | 72 °C | |
| Final extension | 2 minutes | 72 °C | 1 |
| Hold | ∞ | 1 5°C |
Table 2: PCR cycling conditions
| Components | Unmodified RNA | me1Ψ-UTPmodified RNA |
| RNase-Free Water | variable | variable |
| Linearized template DNA with T7 RNAP promoter | variable (1 µg) | variable (1 µg) |
| 10x T7 TranscriptionBuffer | 2 µL | 2 µL |
| 100 mM N1-methyl Pseudouridine | – | 1.5 µL |
| 100 mM ATP | 1.8 µL | 1.8 µL |
| 100 mM UTP | 1.8 µL | – |
| 100 mM CTP | 1.8 µL | 1.8 µL |
| 100 mM GTP | 1.8 µL | 1.8 µL |
| 100 mM DTT | 2 µL | 2 µL |
| 40 U/µL RNase Inhibitor | 0.5 µL | 0.5 µL |
| T7 Enzyme Solution | 2 µL | 2 µL |
| Total Reaction Volume | 20 µL | 20 µL |
Table 3: IVT reaction mixture preparation
| Components | RNA ladder | RNA sample |
| 2x RNA loading dye (NEB) | 6 µL | 6 µL |
| RNA | 2 µL | 1 µL (0.5-1 µg ) |
| DEPC treated water | 4 µL | 5 µL |
| Total volume | 12 µL | 12 µL |
Table 4: RNA loading dye preparation
| Components | Quantity |
| 10x CappingBuffer | 10 µL |
| 20 mM GTP | 5 µL |
| 20 mM SAM | 2.5 µL |
| RNase Inhibitor | 2.5 µL |
| 2'-O-Methyltransferase | 4 µL |
| Total Volume | 24 µL |
Table 5: Enzymatic Cap-1 synthesis reaction mixture
| Components | Quantity |
| 5’-Capped IVT RNA | 100 µL |
| RNase Inhibitor | 0.5 µL |
| 10x A-PlusTailingBuffer | 12 µL |
| 20 mM ATP | 6 µL |
| 4 U/µL A-Plus Poly(A) Polymerase | 5 µL |
| Total Volume | 123.5 µL |
Table 6: Poly A tailing reaction mixture
| Charge ratio | Liposome | DI water | mRNA(500ng) | DI water |
| 1:1 | 1.5 μL | 8.5 μL | 1 μL | 9 μL |
| 2:1 | 3 μL | 7 μL | 1 μL | 9 μL |
| 4:1 | 6 μL | 4 μL | 1 μL | 9 μL |
| 8:1 | 12 μL | – | 1 μL | 9 μL |
| Total volume (20 μL) | 10 μL | 10 μL | ||
Table 7: Preparation of lipoplex based on charge ratios
| Name | Components |
| 50x TAE buffer | Dissolve 50 mM EDTA sodium salt, 2 M Tris, 1 M glacial acetic acid in 1 L of water |
| HEK-293T and NIH/3T3 cell culture medium | DMEM with 4.5 g/L glucose, L-glutamine, 1% penicillin/streptomycin and 10% FBS |
Table 8: Preparation of buffer and media
