Protoplast isolation
This protocol is effective for the protoplast sorting of fluorescent A. thaliana root marker lines. These markers lines have been developed by the fusion of fluorescent proteins with genes expressed specifically in target cell types, or using enhancer trap lines (Figure 1). Numerous tissues and organs have been dissected into cell types expressing specific fluorescent markers in model plants and crops.
FACS population, sorted cells, and library QC
By using a wild-type plant as a control and setting gates such as forward scatter (FSC) and side scatter (SSC), we determined a major population of cells of interest and a baseline for autofluorescence and successfully sorted the fluorescence-specific marked cells (Figure 2). The quality of the final sequencing libraries (from step 4.15) was determined by the fragment size distribution analysis. The representative results of the RNA-seq library of about 2,000 xylem-pole pericycle cells, lateral root primordia cells, endodermis/cortex cells, and lateral root cap cells are shown in Figure 3A–D.
Analysis of expression patterns
The quality of the sequencing data can be evaluated from multiple analysis procedures, such as sequencing depth, mapping rate, and fastQC reports. The accuracy and sensitivity of the sequencing data can be demonstrated by the presence of a series of cell-type enriched genes, which can be identified from the DEG analysis between isolated cell types and the whole tissue. Genes with significantly higher expression levels in certain cell types can be identified as cell-type enriched genes. Meanwhile, the genome-browser views of each cell type can be compared side by side to show the expression levels of known marker genes and test whether the expression pattern of the marker genes can be reconstructed in the cell-type expression data. As an example, the genes that are enriched in any of the four root cell types were clustered and shown in heatmap, which showed the specificity of gene expression among different cell types (Figure 4). YUCCA3, MYB36, WOX5, and PFA1 were examined, and the expression patterns were as expected according to earlier reports21,22,23,24. Data analysis pipelines and representative raw sequencing data are available in a public repository (github.com/gaolabsjtu/root_cell_types_RNAseq).
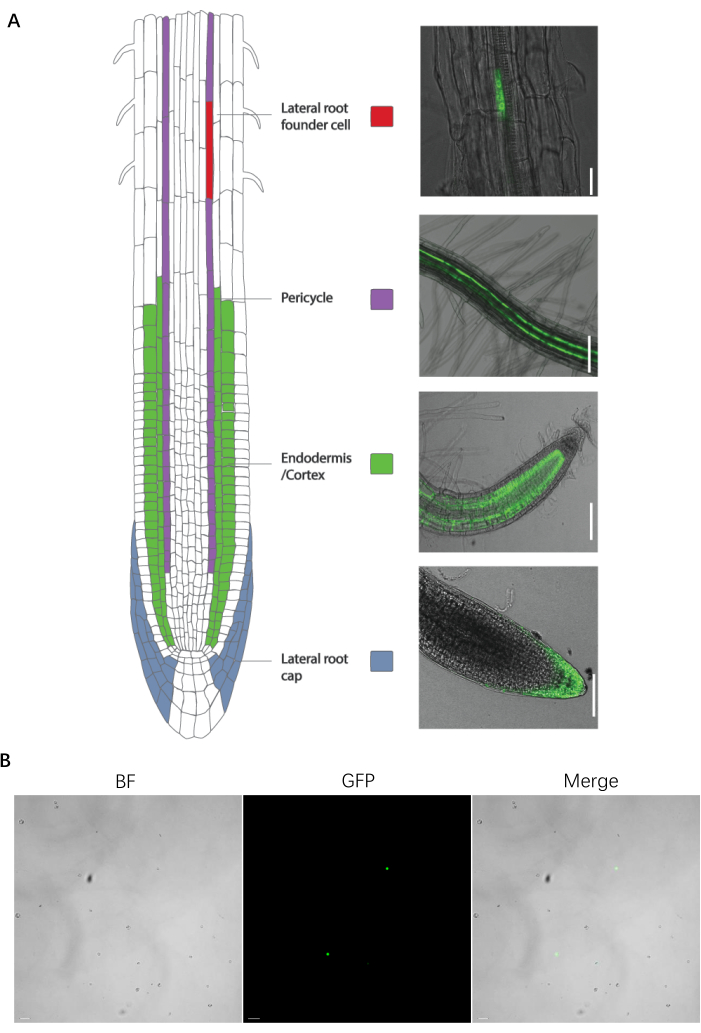
Figure 1: The specific cell type marker line of root and protoplast preparation. (A, left) Enhancer trap line introduction and images. (A, right) The specific cell type marker line of the root. (B) An image of the protoplast preparation before sorting. The scale bars for the lateral root founder cell, pericycle, endodermis/cortex, lateral root cap, and protoplasts represent 25 µm, 100 µm, 100 µm, 75 µm, and 20 µm, respectively. Please click here to view a larger version of this figure.
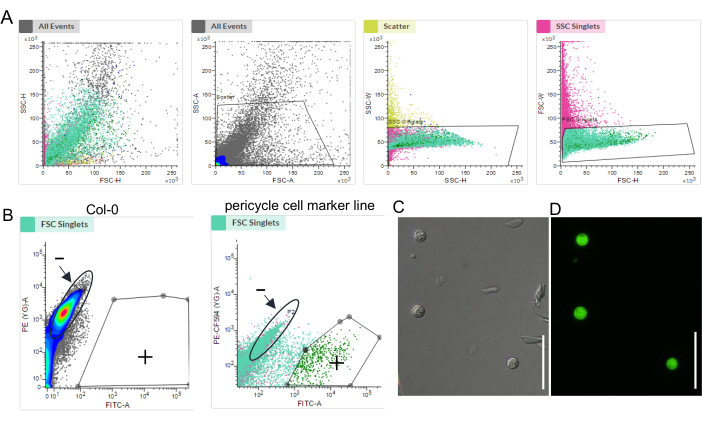
Figure 2: Establishment of FACS for specific root cell types. Example of a FACS experiment: (A) the major population using SSC and FSC; (B) the GFP-positive (+) and GFP-negative (−) gates; (C) the brightfield and (D) fluorescence images of the sorted cells. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
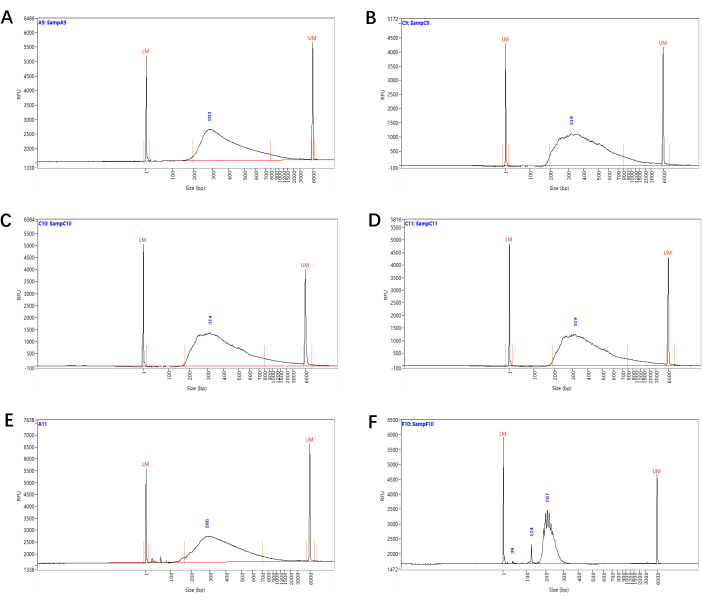
Figure 3: Size distribution of Smart-seq2 sequencing libraries. Representative fragment size distributions of the sequencing libraries (from step 4.15) generated from xylem-pole pericycle cells (A), lateral root primordia cells (B), endodermis/cortex cells (C) and lateral root cap cells (D). (E) A small-size library with primer/adapter dimer peaks, which can still be sequenced after size selection. (F) A library with an abnormal size distribution, which indicated unsuccessful library preparation. Please click here to view a larger version of this figure.
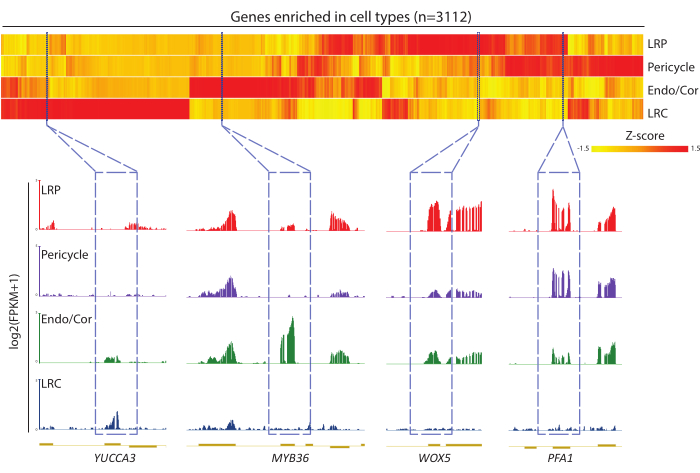
Figure 4: Cell-type-enriched gene analysis. A DEG analysis was carried out between each cell type and the whole root; genes with more than four-fold upregulation in each cell type were identified as cell-type-enriched genes. These genes were combined, clustered, and visualized in the heatmap (upper panel) plot. The cell-type-enriched genes included many known marker genes; typical marker genes such as YUCCA3, MYB36, WOX5, and PFA1 were chosen to show in the genome browser (lower panel). Abbreviations: LRP = lateral root primordia; Endo/Cor = endodermis/cortex; LRC = lateral root cap. Please click here to view a larger version of this figure.
| Components | Volume (µL) |
| 10% Triton X-100 | 0.33 |
| RNase inhibitor | 0.55 |
| DTT (0.1 M) | 0.22 |
Table 1: Reaction components for the preparation of the cell lysis buffer (mixture A).
| Components | Volume (µL) |
| Oligo-dT30VN (100 µM) | 0.44 |
| dNTPs (10 mM) | 4.4 |
Table 2: Reaction components for the preparation of mixture B.
| Components | Volume (µL) |
| SuperScript IV buffer (5x) | 8.8 |
| Betaine (5 M) | 8.8 |
| DTT (0.1 M) | 2.2 |
| MgCl2 (1 M) | 0.264 |
| TSO (100 µM) | 0.44 |
| SuperScript IV reverse transcriptase (200 U/µL) | 2.2 |
| RNase inhibitor | 1.1 |
Table 3: Reaction components for the preparation of the reverse transcription PCR mixture (mixture C).
| Cycle | Temperature (°C) | Time |
| 1 | 50 | 90 min |
| 2-11 | 55 | 2 min |
| 50 | 2 min | |
| 12 | 70 | 15 min |
| 13 | 4 | ∞ |
Table 4: Reverse transcription (RT) PCR settings for synthesizing cDNA from mRNA.
| Components | Volume (µL) |
| KAPA HiFi HotStart ReadyMix (2x) | 44 |
| IS PCR primer (10 µM) | 0.88 |
Table 5: Reaction components for the preparation of the pre-amplification reaction mixture (mixture D).
| Cycle | Temperature (°C) | Time |
| 1 | 98 | 5 min |
| 2-13 | 98 | 20 s |
| 67 | 30 s | |
| 72 | 3 min | |
| 14 | 72 | 5 min |
| 15 | 4 | ∞ |
Table 6: PCR program settings for the pre-amplification reaction.
