1. Starting Material and Expected Yields
Following 1 hour (hr) of 4sU-exposure newly transcribed RNA represents about 1 – 4% of total cellular RNA. This will be lower in growth-arrested cells as they no longer synthesize RNA to account for cell growth/replication. When labeling for 1 hr, we recommend starting the assay with 60 – 80 μg of total RNA. Starting with less than 30 μg of total RNA results in small RNA pellets that are hard to see after the biotinylation step and thus may be easily lost. Input RNA levels may be increased to as much as 150 μg for very short durations of labeling (e.g. 5 – 10 min). When the duration of RNA labeling is shortened from 1 hr to 5 min the contribution of short-lived intronic sequences in newly transcribed RNA increases from ~60% to ~80% 9. As introns are substantially longer compared to coding sequences as well as 5′- and 3′-UTRs, the amount of newly transcribed RNA, which can be purified following short- or even ultra-short 4sU-tagging, does not drop linearly. As such, we obtained >0.5% of total RNA following 5 min of 4sU-tagging in non-adherent human B-cell lines 9. It should, however, be noted that higher concentration of 4sU and slightly longer durations of labeling may be required to achieve similar 4sU incorporation rates in adherent cells. While even a low 4sU-incorporation rate will allow efficient capture and purification of large, uridine-rich transcripts, very short transcript with low uridine content (e.g. miRNAs) are likely to escape purification even when using high 4sU concentrations (>1 mM). In NIH-3T3 murine fibroblasts, 1 hr of 200 μM 4sU exposure labeled newly transcribed RNA with about one 4sU residue per 50 – 100 nucleotides (nt) 5. This should allow highly efficient recovery of transcripts >500 – 1000 nt in length. Accordingly, we only observed a minor transcript size bias when labeling for 1 hr using 200 μM 4sU in both murine fibroblasts and human B-cells 7. While 1 hr of 200 μM 4sU did not result in any significant alterations in cellular transcripts levels in murine fibroblasts, prolonged exposure of cells to ≥200 μM 4sU does result in a measurable growth deficit within 24 hr (unpublished data). Therefore, both the duration of labeling and the employed 4sU-concentration should be minimized to avoid ectopic or toxic effects. An easy way to determine the minimal 4sU-concentration required for efficient recovery of newly transcribed RNA is to purify newly transcribed RNA following 4sU-labeling with increasing concentrations of 4sU (e.g. 50 – 1600 μM). As shown in Figures 2A and 2B, recovery of newly transcribed RNA labeled for 1 hr in primary human fibroblasts increased substantially from 50 to 200 μM 4sU but then started to plateau.
2. Dot Blot Quantification of 4sU Incorporation (optional)
In some cases it may be of interest to measure the amount of 4sU incorporation in total RNA. This is best done by dot blot analysis on the biotinylated RNA using a streptavidin conjugate. Due to its chemical nature iodoacetyl-biotin is more reactive to thiol-groups than biotin-HPDP resulting in the biotinylation of virtually all 4sU residues in newly transcribed RNA. It is important to note that, like biotin-HDPD, iodoacetyl-biotin is not water-soluble and is thus efficiently removed by chloroform extraction as performed for biotin-HPDP. Therefore, identical reaction conditions and concentrations can be employed as when using biotin-HPDP. However, iodoacetyl-biotin is not reversible. It can thus not be used for the purification of newly transcribed RNA in column based approaches. While the use of iodoacetyl-biotin allows to quantify 4sU-incorporation, biotin-HPDP based measurements consider both 4sU-incorporation and biotinylation efficiency. Employing the two biotinylation reagents to the same sample allows for the measurement of the biotinylation efficiency of RNA-incorporated 4sU. Biotinylation efficiency of biotin-HPDP for 4sU-labeled RNA seems to be approximately three fold less than that of iodoacetyl-biotin indicating that only about one in three 4sU residues in newly transcribed RNA is actually biotinylated by biotin-HPDP(Figure 3). By comparing the sample signal intensities with the biotinylated control DNA oligo, biotinylation densities can be measured. For most mammalian cell lines a positive signal should still be detectable in 10 ng of biotinylated RNA following 1 hr of 200 μM 4sU labeling. A weak background signal is usually detectable for the highest concentration (1 μg) of unlabeled RNA.
3. Purification of Newly Transcribed RNA
Recovery of newly transcribed RNA is highly quantitative. If you started with the same RNA concentration you can expect to obtain the same amounts of newly transcribed RNA for all samples. Like many column-based assays, collection of newly transcribed RNA using the RNeasy MinElute kit may result in additional absorption at 230 – 260 nm (presence of detergents derived from the washing buffers) which may interfere with OD260 measurements. This is seen to a lesser extent when using a fresh 2 ml collection tube for each centrifugation step. Nevertheless, any unreasonably high OD measurements (>2-fold greater than other samples) should be considered with care, particularly if OD260/280 ratios are <1.7. For down-stream analyses it is thus often best to use the same amount of template RNA volume for all samples. In cases where the yields of labeled RNA are lower than expected check for signs of RNA degradation by electrophoretical analysis. Newly transcribed RNA contains significantly greater amounts of large, unspliced transcripts with the typical rRNA bands being much less prominent (Figure 4).
4. Quantification of Newly Transcribed RNA
Finally, incorporation rates of 4sU in newly transcribed RNA can be directly quantified by spectrophotometric analysis based on the absorption maximum of 4sU at 330 nm and the OD330/260 ratio 5,18. This requires >3 μg of labeled RNA concentrated in a small volume (10 – 20 μl) by isopropanol/ethanol precipitation. To avoid losing the small RNA pellet co-precipitation with 30 μg of nuclease-free glycogen (Fermentas, #R0551) should be performed. An additional peak is visible at 330 nm reflecting the incorporation rate of 4sU into newly transcribed RNA (Figure 5).

Figure 1. Principle of metabolic labeling with 4-thiouridine (4sU). 4sU is added to cells for the required (5 – 120 min) time followed by preparation of total cellular RNA. Following thiol-specific biotinylation, total cellular RNA is separated into 4sU-labeled, newly transcribed RNA, and unlabeled, pre-existing RNA using streptavidin-coated magnetic beads. Newly transcribed RNA is recovered from the beads using a reducing agent which cleaves the disulfide bonds that link the newly transcribed RNA to the beads. Click here to view larger figure.
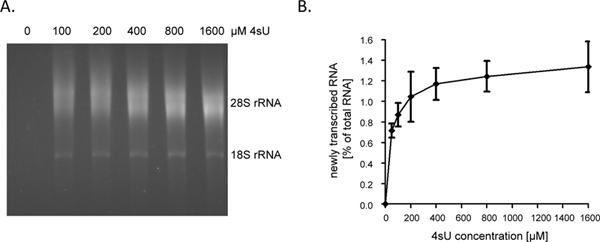
Figure 2. Recovery of newly transcribed RNA following increasing concentrations of 4sU. (A) Primary human foreskin fibroblasts (HFF) were incubated with 100, 200, 400, 800 or 1,600 μM of 4sU. Newly transcribed RNA was purified from 50 μg total cellular RNA and subjected to electrophoretical analysis. As expected, a concentration dependent increase in recovered newly transcribed RNA was observed which started to plateau at higher concentrations. (B) Amounts of purified newly transcribed RNA were quantified using the ImageJ 1.45s software. Combined data of four independent experiments on the amounts of newly transcribed RNA recovered following different concentrations of 4sU-labeling ranging from either 50 – 800 μM 4sU (n = 2) or 100 – 1,600 μM 4sU (n = 2) are shown. Click here to view larger figure.
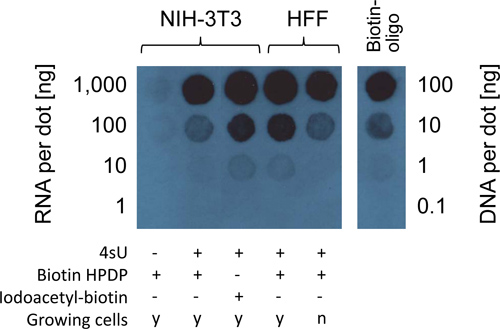
Figure 3. Estimation of 4sU incorporation in 4sU-labeled total RNA using dot blot analysis. Total RNA was isolated from NIH-3T3 murine fibroblasts or human foreskin fibroblasts (HFF) incubated with 200 μM 4sU for one hour. No 4sU was added to one dish as negative control. For HFF both contact inhibited (n = non-growing cells) and growing cells (y) were included. RNA was isolated using Trizol reagent and subsequently conjugated to biotin-HPDP or iodoacetyl-biotin. Concentration of each sample was adjusted to 200 ng/μl and 5 μl of this dilution (i.e. 1 μg of RNA), as well as three subsequent 10-fold dilutions (i.e. 100, 10, and 1 ng RNA, respectively), were all spotted on a piece of Zeta membrane. 5 μl dilutions of biotin-labeled DNA oligo were placed on the membrane as positive controls at concentrations ranging from 20 ng/μl down to 20 pg/μl (i.e. 100 to 0.1 ng, respectively). Biotin density was probed using a streptavidin-horseradish peroxidase conjugate.
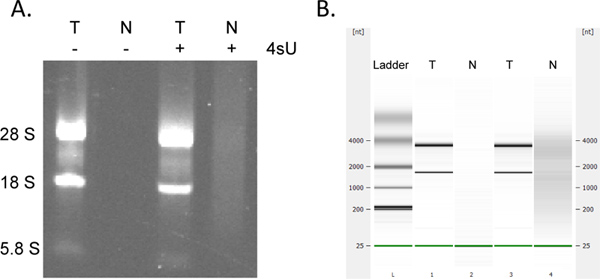
Figure 4. Electrophoretical analysis of newly transcribed and total RNA. Total RNA (T) and newly transcribed RNA (N) prepared from murine NIH-3T3 fibroblasts cultured both in presence and absence of 500 μM 4sU for 1 hr was analyzed by agarose gel electrophoresis (A) and (in the same order) using the Agilent Bioanalyser (B). No RNA was recovered without 4sU treatment of cells. Purified newly transcribed RNA contains greater amounts of high molecular weight mRNAs and significantly less mature rRNAs than total RNA as notable between the 28S, 18S, and 5.8S rRNA bands. Click here to view larger figure.
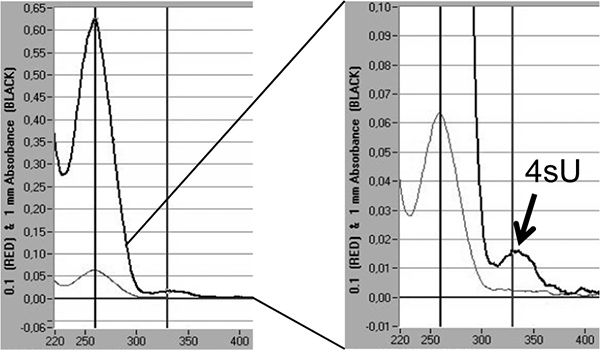
Figure 5. Quantification of 4sU incorporation in newly transcribed RNA by spectrophotometric analysis. Newly transcribed RNA purified from 2 x 100 μg total RNA following 1 hr of 200 μM 4sU in murine NIH-3T3 fibroblasts. Newly transcribed RNA was precipitated with isopropanol/ethanol after adding 30 μg of nuclease-free glycogen. Spectrophotometric analysis of newly transcribed RNA obtained by a Nanodrop 1000 spectrophotometer is shown. The light grey lines represent measurements at 0.1 mm while the thicker, dark grey lines represent measurements at 1 mm fluid column. On the right, a magnification of the peak of extinction representing the incorporated 4sU-residues is shown. Based on the extinction co-efficient of 4sU 18 the incorporation rates of 4sU can be estimated.
| Duration of labeling [min] | Recommended 4sU concentration [μM] |
| 120 | 100 – 200 |
| 60 | 200 – 500 |
| 15 – 30 | 500 – 1000 |
| <10 | 500 – 2000 |
Table 1. Recommended 4sU concentrations.
