Massively Parallel Reporter Assays in kultivierte Säugerzellen
Summary
The genetic reporter assay is a well-established and powerful tool for dissecting the relationship between DNA sequences and their gene regulatory activities. Coupling candidate regulatory elements to reporter genes that carry identifying sequence tags enables massive parallelization of these assays.
Abstract
The genetic reporter assay is a well-established and powerful tool for dissecting the relationship between DNA sequences and their gene regulatory activities. The potential throughput of this assay has, however, been limited by the need to individually clone and assay the activity of each sequence on interest using protein fluorescence or enzymatic activity as a proxy for regulatory activity. Advances in high-throughput DNA synthesis and sequencing technologies have recently made it possible to overcome these limitations by multiplexing the construction and interrogation of large libraries of reporter constructs. This protocol describes implementation of a Massively Parallel Reporter Assay (MPRA) that allows direct comparison of hundreds of thousands of putative regulatory sequences in a single cell culture dish.
Introduction
Massively Parallel Reporter Assays (MPRA) erlauben Multiplex-Messung der Transkriptionsregulationsaktivitäten Tausende bis Hunderttausende von DNA-Sequenzen 1-7. In ihrer allgemeinsten Ausführung wird durch Multiplexen Koppeln jeder Sequenz von Interesse ein synthetisches Reportergen, das eine Identifizierungssequenz Tag stromabwärts eines offenen Leserasters enthält erzielt (ORF Abbildung 1). Nach der Transfektion der RNA-Isolierung und Sequenzierung des Tief 3 'Enden der Reportergen-Transkripten, können die relativen Aktivitäten der gekoppelten Sequenzen aus der relativen Häufigkeit der Ermittlung Variablen abgeleitet werden.
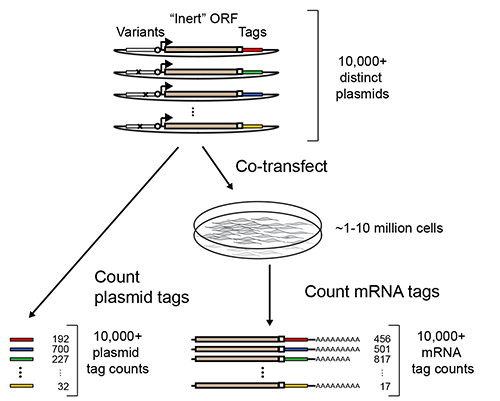
Abbildung 1. Übersicht über MPRA. Eine Bibliothek von MPRA Reporterkonstrukte durch Kopplung mutmaßlichen regulatorischen Sequenzen zu synthetischen gebautReportergene, die aus einem "inerten" ORF bestehen (wie GFP oder Luciferase), gefolgt von einem identifizierenden Sequenz-Tag. Die Bibliothek wird in Massen in eine Population von kultivierten Zellen transfiziert und transkribiert Reporter mRNA wird anschließend zurückgewonnen. Deep sequencing wird verwendet, um die Anzahl der Vorkommen der einzelnen Tag unter den Reporter-mRNAs und den transfizierten Plasmiden zählen. Das Verhältnis der mRNA zählt über Plasmid Zahlen können verwendet werden, um die Aktivität der entsprechenden Regulationssequenz abzuleiten. Angepasst mit Erlaubnis von Melnikow, et al 2.
MPRA kann zu einer breiten Vielzahl von experimentellen Designs angepasst werden, einschließlich 1) umfassende Mutagenese einzelner Gen regulatorische Elemente, 2) Überprüfung auf neue regulatorische Elemente in einem Ort von Interesse, 3) Testen der Wirkung der natürlichen genetischen Variation in einer Menge von putativen Promotoren, Enhancer oder Silencer, und 4) semi-rationale Konstruktion von synthetischen regulatorische Elemente. Libibliotheken von Sequenzvarianten können unter Verwendung einer Vielzahl von Verfahren, einschließlich Oligonukleotid-Synthese von Bibliotheken (OLS) auf programmierbaren Mikroarrays 2,3,6,7, Montage von degenerierten Oligonukleotiden 1,4, kombinatorische Ligations 8 und Fragmentierung der genomischen DNA, 5 erzeugt werden.
Dieses Protokoll beschreibt die Konstruktion einer Bibliothek von Promotorvarianten mit OLS und die pMPRA1 und pMPRAdonor1 Vektoren (Addgene IDs 49349 und 49352 sind; http://www.addgene.org) transiente Transfektion dieser Bibliothek in kultivierte Säugerzellen und die anschließende Quantifizierung der Promotor-Aktivitäten von deep sequencing der ihnen zugeordneten Tags (Tag-Seq). Frühere Versionen von diesem Protokoll wurden in der Forschung in Melnikov et al. Nature Biotechnology 30, 271-277 (2012) und in Kheradpour et al. Genome Research 23, 800-811 (2013) berichtet, verwendet.
Protocol
Representative Results
Discussion
MPRA is a flexible and powerful tool for dissection of sequence-activity relationships in gene regulatory elements. The success of MPRA experiments depend on at least three factors: 1) careful design of the sequence library, 2) minimization of artifacts during amplification and cloning, and 3) high transfection efficiency.
The possible lengths of the variable regions in the reporter constructs are largely determined by the synthesis or cloning technology used. Standard OLS is generally limited to about 200 nt, but this protocol is compatible with inserts up to at least 1,000 nt. Note that variable regions that are highly repetitive or contain strong secondary structures may end up underrepresented due to PCR and cloning biases. The length of the tags that identify each of the variable regions should be 10-20 nt and the collection of tags should ideally be designed such there are at least two nucleotide differences between any pair. Tags that contain the seed sequences of known microRNA or other factors that might influence mRNA stability should also be avoided when possible.
A key parameter in the design of MPRA experiments is the total number of distinct reporter constructs to be included in the library (the design complexity, denoted CD). In practice, CD is limited by the number of cultured cells that can be transfected. As a rule of thumb, the total number of transfected cells should be at least 50-100 times greater than CD. For example, if 20 million cells can be transfected with a transfection efficiency of 50%, then CD should be no more than ~200,000. Note that CD is equal to the number of distinct regulatory sequence variants multiplied by the number of distinct tags per sequence. The more distinct tags are linked to each regulatory sequence, the more accurate the estimate of the activity of that sequence can be made (because measurements from distinct tags can be averaged), but the fewer distinct variants can be assayed in one experiment. The optimal choice depends on the experimental design. In a simple “promoter bashing” experiment, where a mathematical model will be fitted to the aggregated measurements, a single tag per variant is usually sufficient. In a screen for single-nucleotide polymorphisms that cause changes in regulatory activities, it may be necessary to use 20 or more tags per allele to obtain statistically robust results, because comparing each pair of alleles requires a separate hypothesis test.
If the sequences to be assayed are not expected to contain transcription start sites, a constant promoter can also be added in the same fragment. For example, pMPRAdonor2 (Addgene ID 49353) includes a minimal TATA-box promoter that is useful when the upstream variable region is expected to have significant enhancer activity, while pMPRAdonor3 (Addgene ID 49354) includes a modified, strong SV40 viral promoter that is useful when the variable region is expected to contain silencer activity or other negative regulatory elements.
Raw OLS products often contain a significant fraction of truncated oligonucleotides. These may interfere with accurate PCR amplification of the designed sequences, particularly when there is significant homology between them. Using PAGE purification to remove truncated synthesis products and emulsion PCR to minimize amplification artifacts are effective techniques for ensuring high library quality. If either step is impractical, it is imperative to minimize the number of PCR cycles used at each amplification step. Selection and expansion of the cloned library in liquid culture is generally sufficient to maintain the design complexity, but if recombination-prone vectors are to be used or significant representation bias is observed, the recovered cells can instead be plated directly onto large LB agar plates, expanded as individual colonies and then scraped off for DNA isolation. It is also important to consider the potential impact of synthesis errors, which are typically found at a rate of 1:100-500 in OLS. Full-length sequencing of the reporter constructs prior to transfection is recommended to identify and correct for such errors.
It is not necessary to introduce reporter constructs into every cell in the transfected culture, but transfection efficiencies below ~50% may lead to poor signal to noise ratios. It is advisable to optimize transfection conditions prior to performing MPRA experiments in a new cell type. When working with hard-to-transfect cell types, MPRA signals can be boosted by pre-selecting transfected cells. The pMPRA vector series includes variants that constitutively express a truncated cell surface marker that can be used to physically enrich for transfected cells prior to RNA isolation (for example, Addgene IDs 49350 and 49351).
Declarações
The authors have nothing to disclose.
Acknowledgements
Diese Arbeit wurde von der National Human Genome Research Institute der National Institutes of Health unter Preis Anzahl R01HG006785 unterstützt.
Materials
| Oligonucleotide library synthesis | Agilent, CustomArray or other OLS vendors | custom | If using OLS construction method |
| pMPRA1 | Addgene | 49349 | MPRA plasmid backbone |
| pMPRAdonor1 | Addgene | 49352 | luc2 ORF donor plasmid |
| TE 0.1 Buffer (10 mM Tris-HCl, 0.1 mM EDTA, pH 8.0) | Generic | n/a | OLS buffer |
| Novex TBE-Urea Gels, 10% | Life Technologies | EC6875BOX | PAGE purification of OLS products |
| Novex TBA-Urea Sample Buffer | Life Technologies | LC6876 | PAGE purification of OLS products |
| SYBR Gold Nucleic Acid Gel Stain | Life Technologies | S-11494 | PAGE purification of OLS products |
| Micellula DNA Emulsion & Purification Kit | EURx/CHIMERx | 3600-01 | Library amplification by emulsion PCR |
| Herculase II Fusion DNA Polymerase | Agilent | 600675 | Polymerase for emulsion PCR |
| SfiI | New England Biolabs | R0123S | Library cloning with pMPRA vectors |
| KpnI-HF | New England Biolabs | R3142S | Library cloning with pMPRA vectors |
| XbaI | New England Biolabs | R0145S | Library cloning with pMPRA vectors |
| T4 DNA Ligase (2,000,000 units/ml) | New England Biolabs | M0202T | Library cloning with pMPRA vectors |
| One Shot TOP10 Electrocomp E. coli | Life Technologies | C4040-50 | Library cloning with pMPRA vectors |
| LB agar and liquid media with carbenicllin | Generic | n/a | Growth media for cloning |
| E-Gel EX Gels 1% | Life Technologies | G4010-01 | Library verification and purification |
| E-Gel EX Gels, 2% | Life Technologies | G4010-02 | Library verification and purification |
| MinElute Gel Extraction Kit | Qiagen | 28604 | Library and backbone purification |
| EndoFree Plasmid Maxi Kit | Qiagen | 12362 | Library DNA isolation |
| Cell culture media | n/a | n/a | Experiment-specific |
| Transfection reagents | n/a | n/a | Experiment-specific |
| MicroPoly(A)Purist Kit | Life Technologies | AM1919 | mRNA isolation |
| TURBO DNA-free Kit | Life Technologies | AM1907 | Plasmid DNA removal |
| SuperScript III First-Strand Synthesis System | Life Technologies | 18080-051 | cDNA synthesis |
| PfuUltra II Hotstart PCR Master Mix | Agilent | 600850 | Polymerase for Tag-Seq PCR |
| Primers (see text) | IDT | custom | PAGE purify Tag-Seq primers |
Referências
- Kinney, J., Murugan, A., Callan, C. G., Cox, E. C. Using deep sequencing to characterize the biophysical mechanism of a transcriptional regulatory sequence. Proceedings of the National Academy of Sciences USA. 107 (20), 9158-9163 (2010).
- Melnikov, A., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nature Biotechnology. 30 (3), 271-277 (2012).
- Patwardhan, R. P., Lee, C., Litvin, O., Young, D. L., Pe’er, D., Shendure, J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nature Biotechnology. 27 (12), 1173-1175 (2009).
- Patwardhan, R. P., et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nature Biotechnology. 30 (3), 265-270 (2012).
- Arnold, C. D., Gerlach, D., Stelzer, C., Boryń, &. #. 3. 2. 1. ;. M., Rath, M., Stark, A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science (New York, N. Y.). 339 (6123), 1074-1077 (2013).
- Kheradpour, P., et al. Systematic dissection of regulatory motifs in 2000 predicted human enhancers using a massively parallel reporter assay. Genome Research. 23 (5), 800-811 (2013).
- White, M. A., Myers, C. A., Corbo, J. C., Cohen, B. A. Massively parallel in vivo enhancer assay reveals that highly local features determine the cis -regulatory function of ChIP-seq peaks. Proceedings of the National Academy of Sciences USA. 110 (29), 11952-11957 (2013).
- Mogno, I., Kwasnieski, J. C., Cohen, B. A. Massively parallel synthetic promoter assays reveal the in vivo effects of binding site variants. Genome Research. 23 (11), 1908-1915 (2013).
- Schütze, T., et al. A streamlined protocol for emulsion polymerase chain reaction and subsequent purification. Analytical Biochemistry. 410 (1), 155-157 (2011).
- Panne, D., Maniatis, T., Harrison, S. C. An atomic model of the interferon-beta enhanceosome. Cell. 129 (6), 1111-1123 (2007).
