Before selecting a sequence to serve as the IPC in qRPA experiments with target (HIV-1) DNA, internal positive control (IPC) candidates are generated and screened for their ability to amplify in qRPA reactions without HIV-1 DNA present. IPC candidates are longer than the target (HIV-1) DNA to prevent IPC formation from out-competing HIV-1 amplicon formation. As shown in Figure 2A, the generation of two C. parvum IPC candidates was verified by the presence of 415 and 435 bp bands using gel electrophoresis. In qRPA reactions, the shorter IPC candidate exhibited little amplification (Figure 2B), while the longer candidate consistently amplified when a total of 104 and 105 copies were present (Figure 2C). Thus, the longer candidate was chosen to be the IPC for HIV-1 qRPA experiments.
Real-time qRPA may be performed using the target of interest alone or using both the target and the IPC. Figure 3 shows an experiment on the thermal cycler using both HIV-1 DNA and the C. parvum IPC. In this experiment, 2.6 x 104 copies of IPC DNA were added to each reaction, a concentration close to the IPC limit of detection, to avoid affecting the limit of detection of HIV-1 target DNA. As demonstrated in Figure 3A, which displays HEX fluorescence data corresponding to real-time HIV-1 DNA generation, the time at which detectable amplification begins is inversely proportional to the initial target concentration. Thus, amplification is apparent earlier for high concentrations of HIV-1 DNA and later for low concentrations of HIV-1 DNA. In contrast, detectable amplification of IPC DNA begins at approximately the same time because the starting IPC concentration is the same in all samples, as shown in Figure 3B, which displays FAM fluorescence data corresponding to IPC DNA generation during the same experiment. The rate of fluorescence generation from the IPC is inversely proportional to the concentration of HIV-1 DNA due to competition during amplification.
With the goal of developing a field-operable fluorescence reader with qRPA reactions, qRPA experiments may be performed on an upright fluorescence microscope with a stage heater and 1-Channel Precision High Stability temperature controller. Figure 4A and 4B display HEX and FAM fluorescence data collected on a microscope. Data collected on the microscope using laser-cut chips demonstrate slight variability in baseline fluorescence and crests and troughs that may be due to photobleaching. However, the script determines the cycle threshold using the rate of change of fluorescence during the initial amplification period, which is unaffected by baseline fluorescence or variability in fluorescence after the initial amplification has occurred. As seen in Figure 4C, the standard curve built from this experiment has an r2 coefficient of 0.990. Notably, the IPC is amplified only for low concentrations of HIV-1 DNA. Although this behavior differs from experiments performed on the thermal cycler, all samples are still classified as valid using this method.
Data from multiple experiments using known target concentrations may be compiled to construct a standard curve, which may then be used to assess assay performance or quantify samples with unknown concentrations. Figure 5 shows the data from five experiments and an exponential standard curve generated using the script “JoVE_qRPA_standard_curve.m”, relating the initial HIV-1 target concentration to the time at which detectable amplification began. Figure 5 used 5 separate experiments, each containing 2 qRPA reactions at each template concentration to build a standard curve. Note that the exponential fit has a high r2 coefficient of 0.959. The standard curve was then used to predict the concentrations of additional samples with known concentrations for assay validation using the script “JoVE_qRPA_validation_and_quantification.m” script. Table 1 shows the quantification results for 5 additional experiments using the standard curve in Figure 5. Note that the algorithm correctly classified all samples containing HIV-1 DNA as positive and 9 out of 10 of the no-target-control samples as negative. In addition, the average predicted concentration was within one order of magnitude of the correct concentration and the standard deviation for predicted concentrations was less than 0.5 log10 (copies per reaction).
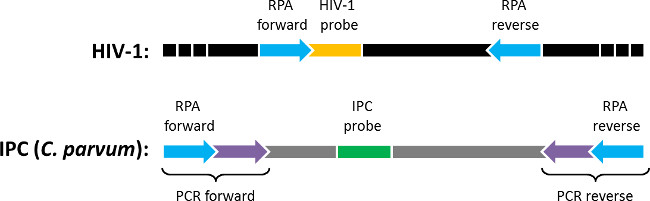
Figure 1: Block diagram of quantitative RPA DNA targets. The qRPA assay amplifies two DNA sequences flanked by identical sequences to which primers bind (“RPA forward” and “RPA reverse”). The amplified sequences are 1) a sequence within the HIV-1 genome (“HIV-1”) that is detected with the “HIV-1 probe” and 2) an internal positive control sequence (“IPC (C. parvum)”) included in each reaction that is detected with the “IPC probe.” The IPC was generated via PCR using the Cryptosporidium parvum genome as a template and primers (“PCR forward” and “PCR reverse”) that contain a region complementary to C. parvum (purple) and a region complementary to HIV-1 (blue). Figure originally published in Analytical Chemistry 2014, reprinted here with permission9.
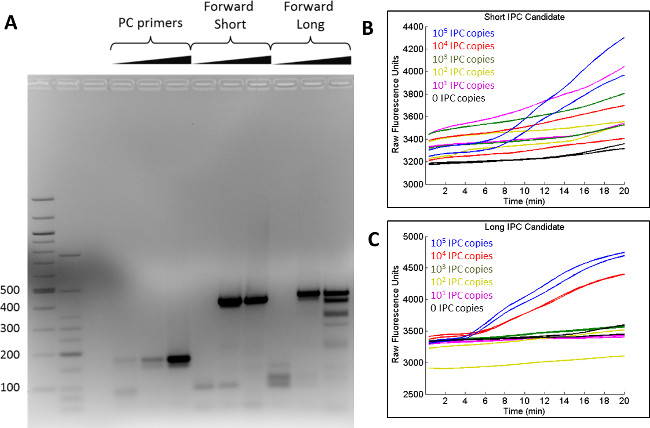
Figure 2: Screening of IPC candidates. (A) Two IPC candidates (‘Forward Short’ and ‘Forward Long’) and a known PCR positive control sequence were generated using PCR and visualized on an agarose gel, where 415 bp and 435 bp bands were visible. (B) The short IPC candidate exhibited little amplification during real-time RPA, while (C) the longer IPC candidate amplified consistently when a total of 104 and 105 copies were present. Figure originally published in Analytical Chemistry 2014, reprinted here with permission9. Please click here to view a larger version of this figure.
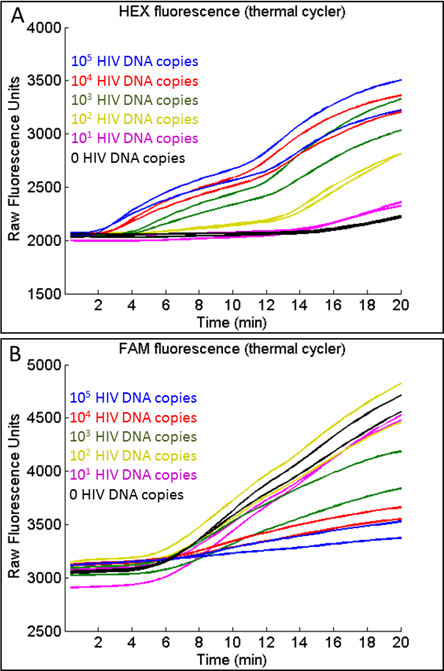
Figure 3: Typical raw fluorescence data generated during co-amplification of HIV-1 DNA and IPC on a thermal cycler. (A) For HIV-1, the onset of detectable amplification, shown by an apparent increase in HEX fluorescence, occurs earlier for high concentrations of HIV-1 DNA. (B) For the IPC, the onset of detectable amplification, shown by an apparent increase in FAM fluorescence, occurs at approximately the same time regardless of the HIV-1 DNA concentration. However, the rate of FAM fluorescence generation is inversely proportional to the amount of HIV-1 DNA present due to competition. Figure originally published in Analytical Chemistry 2014, reprinted here with permission9.
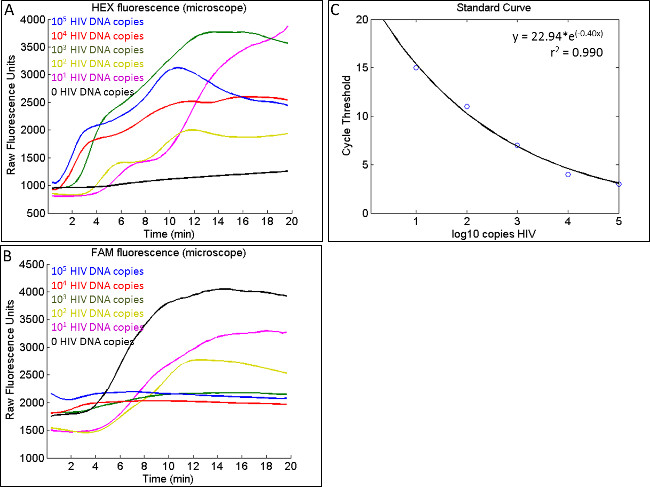
Figure 4: Typical raw fluorescence data generated during co-amplification of HIV-1 DNA and IPC on a microscope. (A) Similar to data generated on the thermal cycler, the onset of detectable HIV-1 amplification, shown by an apparent increase in HEX fluorescence, occurs earlier for high concentrations of HIV-1 DNA. (B) IPC amplification on the microscope, shown by FAM fluorescence, is apparent for low HIV-1 DNA concentrations but not always apparent in samples with high HIV-1 DNA concentrations. (C) A standard curve can be built using the JoVE_standard_curve.m scripts that yields a high r2 coefficient.
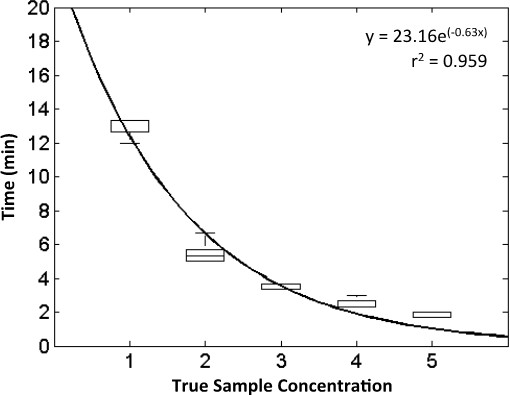
Figure 5: Typical standard curve for HIV-1. Using the JoVE_standard_curve.m script, raw HEX fluorescence data generated during HIV-1 amplification is processed to build a standard curve, which may be used to predict the HIV-1 DNA concentration of unknown samples. This standard curve (z = 1) was generated using data from 5 experiments, in which 6 concentrations were tested using 2 replicates at each concentration. All concentrations are given in log10 copies. Figure originally published in Analytical Chemistry 2014, reprinted here with permission9.
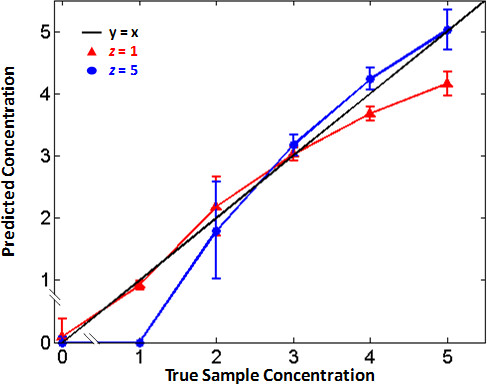
Figure 6: Algorithm tunability. Adjusting the algorithm parameters changes the standard curve used to predict the concentration of unknown samples. The JoVE_validation_and_quantification.m script was used to predict the concentration of unknown samples using z = 1 (red) and z = 5 (blue). All concentrations are given in log10 copies. Predictions are more accurate for low DNA concentrations when z = 1; predictions are more accurate for high DNA concentrations when z = 5. By adjusting the algorithm parameters, the qRPA standard curve may be tuned according to clinical needs. Figure originally published in Analytical Chemistry 2014, reprinted here with permission9.
| Sample Concentration | Average Predicted Concentration | Standard Deviation | Percent of Samples Identified as Positive |
| No target controls | 0.1 | 0.3 | 10% |
| 1 | 0.9 | 0.1 | 100% |
| 2 | 2.2 | 0.5 | 100% |
| 3 | 3 | 0.1 | 100% |
| 4 | 3.7 | 0.1 | 100% |
| 5 | 4.2 | 0.2 | 100% |
Table 1: HIV-1 qRPA assay validation. Using the standard curve in Figure 4, the JoVE_validation_and_quantification.m script was used to predict the concentrations (in log10 copies) of samples for 5 additional experiments. Table originally published in Analytical Chemistry 2014, reprinted here with permission9.
