This protocol describes how to molecularly analyze and visualize polarized membrane biogenesis and lumen morphogenesis in the C. elegans intestine, at the single cell and subcellular level. The twenty-cell single-layered C. elegans intestine is formed by directed cell division and migration during mid embryogenesis. At this time, polarized membrane domains become established, yet de novo polarized membrane biogenesis continues in the mature but expanding epithelium throughout four larval stages until adulthood, allowing to focus the analysis on polarized membrane biogenesis (Figure 1A).
To visualize C. elegans cellular and subcellular components, two strategies are commonly used: immunofluorescence (detailed in this protocol, section 1; Figure 2, Figure 4D-F) and the expression of fluorescence fusion proteins (detailed in the accompanying paper on excretory canal polarized membrane biogenesis2; Figure 1B, Figure 2, Figure 4, Figure 5C). Double and multiple labeling, combining different labels of each or both methods, can resolve membrane asymmetries such as apical and basolateral membrane domains, and the relation of different subcellular components to each other (Figure 2, Figure 4D-E). The membrane-cytoskeleton linker ERM-1::GFP is shown here as an indicator of apical membrane biogenesis that coincides with lumen morphogenesis in this single-layered epithelium. By using this marker, an array of intestinal apical membrane/lumen biogenesis defects and their causative gene defects can be identified by loss-of-function studies, for instance by unbiased genome-wide screens using RNAi (RNAi approaches adopted to the generation of such phenotypes are described in section 2 of this protocol). Figure 3 and Figure 4 show examples of low-to-moderate magnification images of apical membrane/lumen biogenesis phenotypes acquired by a dissecting fluorescence microscope equipped with a high power objective; and of higher magnification images acquired by a confocal laser scanning microscope (these microscopic approaches are described in sections 3 and 4). As an example of quantifying polarized membrane biogenesis defects, the effects of RNAi with let-767 (encoding a steroid dehydrogenase/3-ketoacyl-CoA reductase) and aps-1 (encoding the sigma subunit of the clathrin AP-1 adaptor) on ERM–1::GFP localization and lumen positioning are shown in Figure 5.
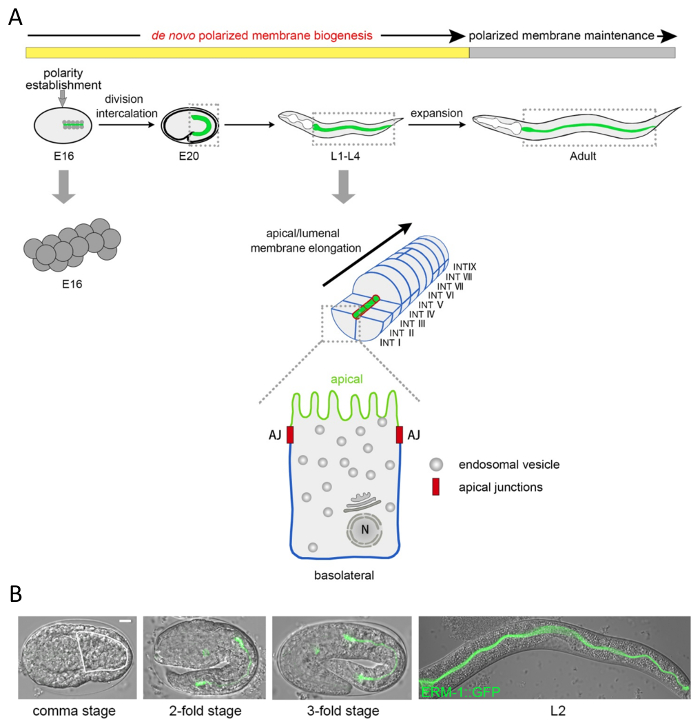
Figure 1: Cellular and subcellular structure and morphogenesis of the wild-type C. elegans intestine. (A) Schematic of C. elegans intestinal development, cellular composition, and endo- and plasma membranes. The C. elegans intestine is generated clonally from the E blastomere born at the 8-cell stage. After four rounds of cell division, its 16 cells (E16 stage) form a radially symmetrical doubled-layered epithelium15. At this stage the cytoplasm of each cell polarizes, with nuclei moving to the future apical, and cytoplasmic components moving towards the opposite (future basal), membrane domains. In one intercalation step left and right ventral cells move (in parallel) into the dorsal cell layer to form the bilaterally symmetrical tube of 9 INT rings. Each cell faces and builds the lumen with its apical/lumenal membrane (green; structurally distinguished by specific membrane microdomains, microvilli) and contacts neighboring cells or the body cavity with its basolateral membranes (blue), except the first INT ring that is formed by four cells. Apical junctions (red) separate apical and basolateral membrane domains. After intercalation, de novo membrane biogenesis continues along with the growth of the intestine during late embryogenesis and the four larval stages into adulthood, where only minimal further growth occurs (phase of polarized membrane maintenance). The magnified single cell indicates the endomembrane system with ER and Golgi above the nucleus (N) and endosomal vesicles. (B) DIC/Nomarski and confocal overlap micrographs of the developing C. elegans intestine labeled with the apical membrane-cytoskeleton marker ERM-1::GFP. The intestine at the comma stage is outlined by a white line (ERM-1::GFP is already faintly expressed at the apical membrane at the beginning of intercalation but cannot be appreciated in this image). Animals here and below are shown with anterior (head) left, posterior (tail) right, dorsal up, ventral down. Scale bar: 5 µm. Please click here to view a larger version of this figure.
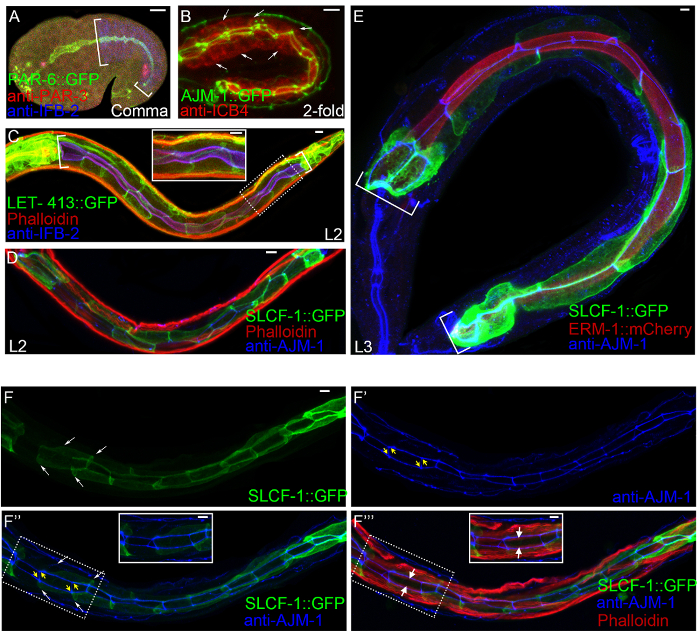
Figure 2: Examples of double and triple labeling of the developing wild-type C. elegans intestine using antibodies and fusion proteins. (A, B) Embryos. (A) Comma stage. PAR-6::GFP (green; component of the apical PAR polarity complex), anti-PAR-3 antibody (red/TRITC (tetramethyl rhodamine isothiocyanate); another component of the apical PAR polarity complex), and MH33 (blue/Cy5 (Cyanine5); anti-IFB-2/intermediate filament). PAR-6::GFP and the PAR-3 antibody label the apical membrane of the C. elegans intestine (bracketed). IFB-2, another apical marker at later stages, is panmembraneously localized at this early stage. PAR-6::GFP and anti-PAR-3 also label the pharynx (left); the intestinal lumen is indicated by their overlap with IFB-2 (turquoise) and the intestinal tube is outlined by blue anti-IFB-2 (right). (B) 2-fold stage. AJM-1::GFP (green; junction component), ICB4 antibody (red/Alexa, ICB4 detects an unknown membraneous intestinal antigen). AJM-1::GFP labels the apical junctions of the C. elegans intestine, visible as peri-lumenal ladder pattern (it also labels hypodermal junctions; not visible since image is focused on the intestine; see section 4, confocal imaging). ICB4 stains all membranes of the C. elegans intestine. Arrows point to basolateral membranes stained by anti-ICB4. (C, D) L2 larvae. (C) LET-413::GFP (green), phalloidin (red/Texas-red, a phallotoxin binding to F-actin) and MH33 (blue/Cyanine5, anti-IFB-2). LET-413/Scribble is a component of the basal polarity complex and localizes to basolateral membranes of the C. elegans intestine (bracketed). Phalloidin and the IFB-2 antibody label the apical submembraneous cytoskeleton of the C. elegans intestine (purple). Phalloidin also strongly stains body wall muscles, overwhelming the intestinal staining. Inset shows higher magnification of boxed area. (E) L3 larva. SLCF-1::GFP (green; integral membrane component/sugar transporter), ERM-1::mCherry (red) and MH27 antibody (blue/Cyanine5, anti-AJM-1). SLCF-1::GFP labels the basolateral membrane, while ERM-1::mCherry labels the apical membrane of the C. elegans intestine (bracketed); AJM-1 labels its apical junctions. (F-F''') L2 larva. (F,F') Single color images. SLCF-1::GFP (green) labels the basolateral membrane (lateral membranes indicated by thin white arrows). MH27 (blue/Cyanine5) labels apical junctions (short yellow arrows). (F'',F''') Overlay images with and without actin. Insets show higher magnification of boxed areas. Note clear distinction of apicolateral angle of intestinal cells by these different membrane/junction markers that appear superficially similar in single color images (F, F'). Thick white arrows in F'''point to the apical/lumenal actin cytoskeleton labeled by phalloidin (otherwise overwhelmed by muscle actin). All images are confocal projections (z-stacks of 0.2 µm), acquired by sequential scanning to avoid bleed-through between channels. Scale bars (for A-E, F-F''' and all insets): 5 µm. Please click here to view a larger version of this figure.
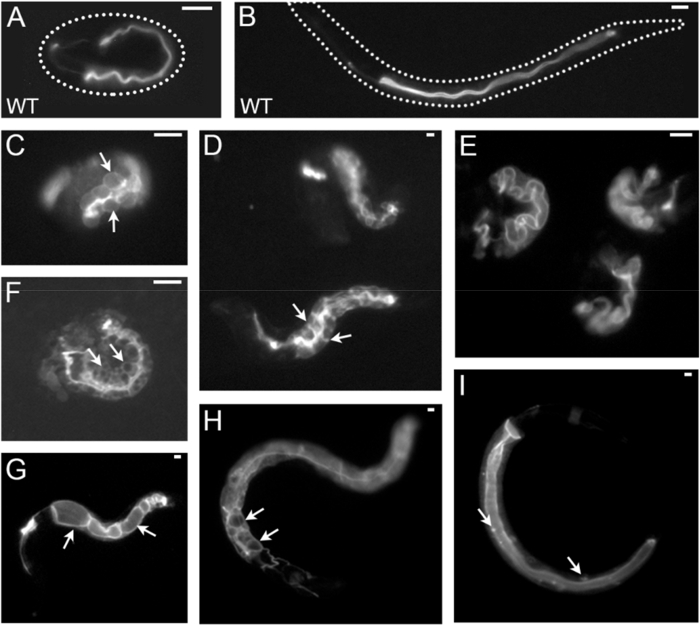
Figure 3: Examples of C. elegans intestinal polarized membrane and lumen morphogenesis defects at low-to-moderate magnification (dissecting fluorescence micrographs). All images are acquired by a dissecting fluorescent microscope equipped with a custom-made high-power stereo fluorescence attachment (Table of Materials). Different magnifications are shown. All phenotypes were obtained by RNAi with different genes in a strain labeled with ERM-1::GFP (localized at intestinal and excretory canal apical/lumenal membranes at embryonic and early larval stages, shown here). The intestinal lumen and polarity phenotypes are: (A, B) Wild type embryo (A) and larva (B); (C,D) basolateral displacement of ERM-1::GFP; intestinal cells are enlarged and appear bloated in this embryo (C, arrows point to single intestinal cells), but are of wild-type size and arrangement in these larvae (D, arrow points to lateral membranes between INT II and III); (E) widened and convoluted lumen in three embryos; (F) ERM-1::GFP broadening into lateral junction area (zigzag lumen) and into intestinal cytoplasm that contains GFP-negative vacuoles (arrows); (G) lumenal cysts inbetween intralumenal adhesions (arrows point to two cysts). (H) Cytoplasmic and basolateral ERM-1::GFP displacement with ectopic lumens (arrows); (I) ERM-1::GFP displacement to GFP-positive puncta (arrows) in the cytoplasm. Excretory canals and excretory canal phenotypes are not described here (canal is shown to the left, intestine to the right in all images). Scale bars: 10 µm. Please click here to view a larger version of this figure.
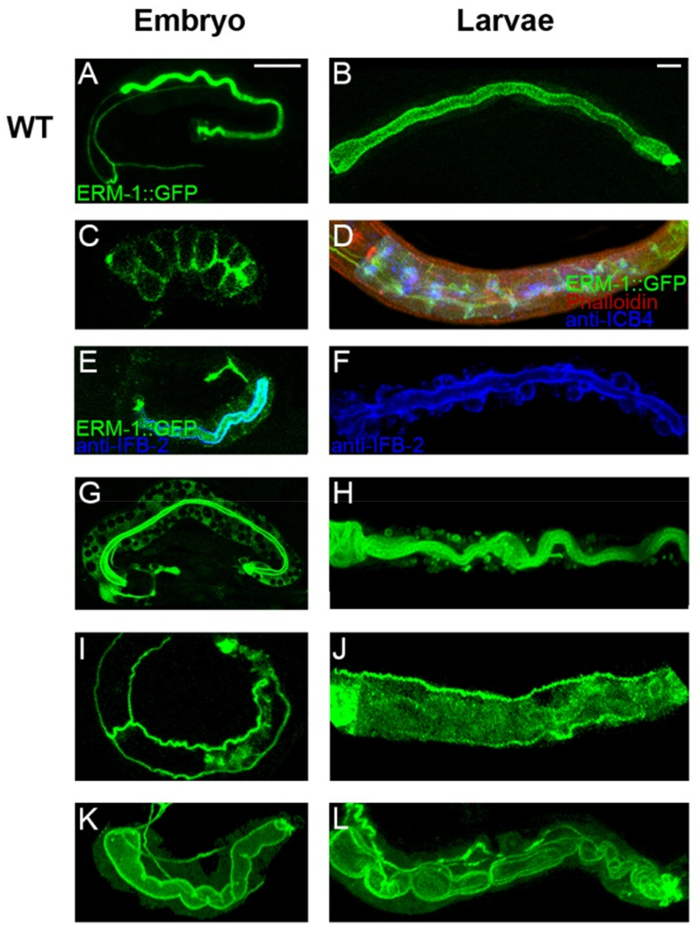
Figure 4: Examples of C. elegans intestinal polarized membrane and lumen morphogenesis defects at higher magnification (confocal images). (A, C, E, G, I, K) Embryos. (B, D, F, H, J, L) Larvae. All phenotypes were obtained by RNAi with different genes in a strain labeled with ERM-1::GFP (green in all images). Imaging is focused on the intestine. Images of embryos also show excretory canals (left side of image), including canal phenotypes (not described). (A, B) Wild type embryo (A) and larva (B). (C) Basolateral displacement of apical ERM-1::GFP (comma stage; late intercalation). (D) Polarity conversion: basolateral displacement of apical ERM-1::GFP and apical accumulation of basolateral ICB4, revealed by double labeling. F-actin (labeled by phalloidin-TRITC) outlines the animals by staining longitudinal muscle bundles (animal is triple labeled). (E) Basolateral displacement of ERM-1::GFP in late 3-fold embryo. The apical IFB-2 antibody (blue/Cyanine5) indicates intact lumen and peri-lumenal intermediate filaments. (F) Ectopic lumens labeled by anti-IFB-2 (blue/Cyanine5). (G) ERM-1::GFP negative vacuoles in intestinal cytoplasm. (H) ERM-1::GFP positive vacuoles in intestinal cytoplasm. (I, J) Absence of lumen in embryo and larvae, respectively. (K) Wide gut. (L) Cystic and convoluted gut. (A, D, H, I, J, K, L) are confocal projections. (B, C, E, F) are confocal sections. Brightness was increased in G to highlight cytoplasmic GFP-negative vacuoles. Scale bars: 10 µm (same for all embryos and larvae, respectively). Please click here to view a larger version of this figure.
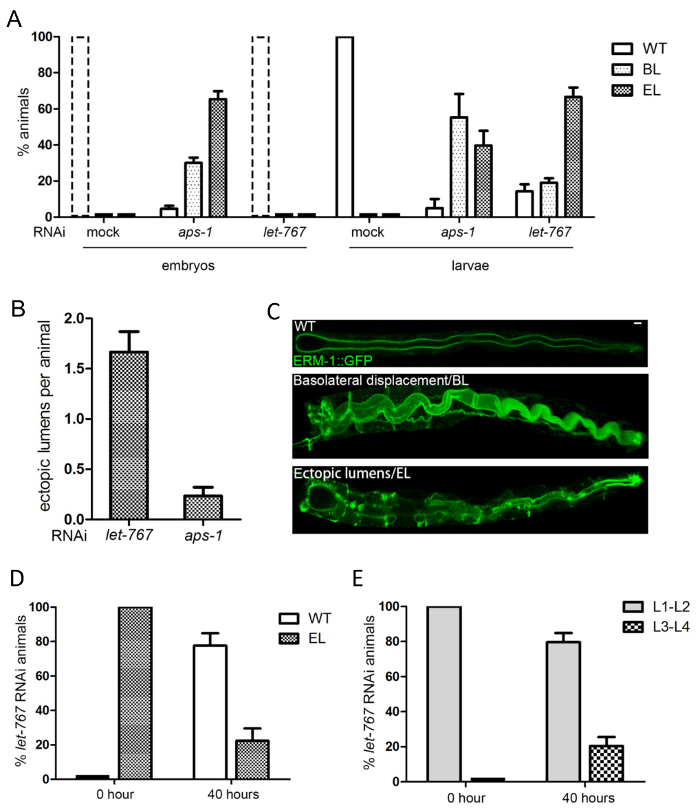
Figure 5: Intestinal polarity conversion and reversion: an example for the quantification of polarized membrane and lumen biogenesis defects. (A, B, C) let-767and aps-1RNAi both cause ERM-1::GFP basolateral displacement (BL) and ectopic lumen formation (EL), but at different developmental stages. (A) Quantification by dissecting microscope: counting of embryos (left) and larvae (right) with polarity phenotypes 2 days after seeding worms on RNAi plates. Note: all mock and let-767(RNAi) animals have hatched at this time (thus there are no embryos, which had, however, all wild-type polarity; broken line columns). aps-1RNAi induces polarity defects already in embryos, whereas let-767RNAi induces them in larvae. The higher percentage of ectopic lumens (vs basolateral displacement) in aps-1RNAi embryos versus larvae is due to arrest of embryos with ectopic lumens. (B) Quantification by confocal microscopy: counting of ectopic lumens per animal at the start of ectopic lumen development in larvae. let-767RNAi induces more ectopic lumens in larvae than aps-1RNAi (aps-1RNAi larvae are "escapers" that have not arrested as embryos). (C) Confocal images for wild-type larvae and larvae with ERM-1::GFP basolateral displacement (BL) and ectopic lumens (EL); scale bar: 5µm. (D, E) Polarity reversion in let-767RNAi animals (counting of animals with and without polarity defect [BL/EL] by dissecting microscope). 20 let-767(RNAi) animals with mild ectopic lumen phenotypes were transferred from an RNAi plate to an OP50 plate on day 4 and evaluated after 40 hours. (D) More than 50% larvae have reverted to wild-type polarity (ERM-1 at the apical membrane). (E) 20% of animals are growing up beyond the L1 larval stage (let-767(RNAi) results in L1 arrest). All data are shown as mean +/- SEM, n = 3. Please click here to view a larger version of this figure.
| Table 1: Examples of markers for the C. elegans larval and adult intestinal membrane system1. | ||||
| Protein name | Subcellular localization | Protein structure/Function | Commercially available C. elegans specific antibodies (DSHB2) | Examples of strains available at the CGC |
| OPT-2/PEPT-1 | apical transmembrane protein | oligopeptide transporter | KWN246 (pha-1(e2123) III, rnyEx133[opt-2(aa1-412):: GFP) + pha-1(+)]) |
|
| AQP-4 | apical transmembrane protein | water channel | ||
| ERM-1 | apical brushborder | membrane–cytoskeleton linker | ERM1 | |
| ACT-5 | apical brushborder | cytoplasmic actin | (3) | |
| IFB-2 | apical brushborder | intermediate filament component | MH33 | |
| EPS-8 | apical brushborder | human-epidermal-growth-factor-receptor-kinase-substrate-8 ortholog | ||
| PAR-6 | apical membrane | apical polarity complex component | ||
| SLCF-1 | basolateral transmembrane protein | monocarboxylate transporter | ||
| AQP-1 | basolateral transmembrane protein | water channel | ||
| LET-413 | basolateral membrane | Scribble homologue, adaptor and polarity determinant | LET413 | |
| HMP-1 | apical junction (CCC4) | α-catenin, cadherin-catenin complex component | FT1609 (unc-119(ed3) III; xnIs528[hmp-1p::hmp-1:: GFP + unc-119(+)]) |
|
| HMR-1 | apical junction (CCC) | E-cadherin, cadherin-catenin complex component | HMR1 | |
| AJM-1 | apical junction (DAC5) | junction integrity molecule, DLG-1/AJM-1complex component | MH27 | SU159 (jcEx44 [ajm-1::GFP + rol-6(su1006)]) |
| DLG-1 | apical junction (DAC) | Discs-large homologue, MAGUK protein, DLG-1/AJM-1complex component | DLG1 | |
| RAB-11 | endosomal vesicles | trafficking6 | RT311 (unc-119(ed3)III; pwIs69 [vha6p::gfp::rab-11, Cbunc-119(+)]) | |
| RAB-5 | endosomal vesicles | trafficking | RT327 (unc-119(ed3)III; pwIs72[pvha6::gfp::rab-5, Cbunc-119(+)]) | |
| RAB-7 | endosomal vesicles | trafficking | RT476 (unc-119(ed3)III; pwIs170[vha6p::gfp::rab-7, Cbunc-119(+)]) | |
| RAB-10 | endosomal vesicles | trafficking | RT525 (unc-119(ed3)III; pwIs206[pvha6::gfp::rab-10 Cbunc-119(+)) | |
| MANS | Golgi | α-mannosidase II | RT1315 (unc-119(ed3)III; pwIs503[pvha6::mans::gfp Cbunc-119(+)]) | |
| 1Examples are selected from resources listed in Table3. | ||||
| 2Developmental Studies Hybridoma Bank. | ||||
| 3Antibodies against vertebrate actin cross-react. | ||||
| 4CCC: cadherin-catenin complex; localizes to the apical part of the apical junction; corresponding to adherens junction (AJ). | ||||
| 5DAC: DLG-1/AJM-1complex; localizes to the basal part of the apical junction; corresponding to tight junction (TJ). | ||||
| 6For additional vesicle-associated molecules expressed in the intestine see ref(22). | ||||
| Note: not all molecules have been tested as fusion proteins under their own promoters or by antibodies. | ||||
| Table 2: Examples of C. elegans intestine-specific promoters and time of expression1. | |||
| Promoters | Expression stage | ||
| elt-2 | expression begins during the 2 E cell stage and persists into adulthood | ||
| vha-6 | expression begins in the late embryo and persists into adulthood | ||
| ges-1 | expression begins at approximately the 4E cell stage and persists into adulthood | ||
| end-1 | expression begins after 1E cell stage and declines during later embryogenesis | ||
| 1Examples are selected from resources listed in Table 3. | |||
| Table 3: Resources to find C. elegans intestine-specific molecules, labeling reagents/strains and antibodies. | |||
| 1. Caenorhabditis Genetics Center (CGC)42 for available reagents and strains | |||
| 2. Wormbase43 for information about intestine-specific molecules, strains and antibodies | |||
| 3. Information about intestine-specific molecules20,44 | |||
| 4. Transgeneome website45 for translational GFP fusion constructs | |||
| 5. C. elegans expression pattern46 for transcriptional GFP fusion constructs | |||
| 6. National BioResource Project (NBRP)::C.elegans47 for information on intestine-specific promoters | |||
| 7. Developmental Studies Hybridoma Bank (DSHB)48 for C. elegans antibodies | |||
| 8. For secondary antibodies and dyes see reference27,28 |
