Three and Four-Dimensional Visualization and Analysis Approaches to Study Vertebrate Axial Elongation and Segmentation
Summary
Here, we describe computational tools and methods that allow visualization and analysis of three and four-dimensional image data of mouse embryos in the context of axial elongation and segmentation, obtained by in toto optical projection tomography, and by live imaging and whole-mount immunofluorescence staining using multiphoton microscopy.
Abstract
Somitogenesis is a hallmark of vertebrate embryonic development. For years, researchers have been studying this process in a variety of organisms using a wide range of techniques encompassing ex vivo and in vitro approaches. However, most studies still rely on the analysis of two-dimensional (2D) imaging data, which limits proper evaluation of a developmental process like axial extension and somitogenesis involving highly dynamic interactions in a complex 3D space. Here we describe techniques that allow mouse live imaging acquisition, dataset processing, visualization and analysis in 3D and 4D to study the cells (e.g., neuromesodermal progenitors) involved in these developmental processes. We also provide a step-by-step protocol for optical projection tomography and whole-mount immunofluorescence microscopy in mouse embryos (from sample preparation to image acquisition) and show a pipeline that we developed to process and visualize 3D image data. We extend the use of some of these techniques and highlight specific features of different available software (e.g., Fiji/ImageJ, Drishti, Amira and Imaris) that can be used to improve our current understanding of axial extension and somite formation (e.g., 3D reconstructions). Altogether, the techniques here described emphasize the importance of 3D data visualization and analysis in developmental biology, and might help other researchers to better address 3D and 4D image data in the context of vertebrate axial extension and segmentation. Finally, the work also employs novel tools to facilitate teaching vertebrate embryonic development.
Introduction
Vertebrate body axis formation is a highly complex and dynamic process occurring during embryonic development. At the end of gastrulation [in the mouse, around embryonic day (E) 8.0], a group of epiblast progenitor cells known as neuromesodermal progenitors (NMPs) become a key driver of axial extension in a head to tail sequence, generating the neural tube and paraxial mesodermal tissues during neck, trunk and tail formation1,2,3,4. Interestingly, the position that these NMPs occupy in the caudal epiblast seems to play a key role in the decision of differentiating into mesoderm or neuroectoderm5. Although we currently lack a precise molecular fingerprint for NMPs, these cells are generally thought to co-express T (Brachyury) and Sox25,6. The exact mechanisms regulating NMP fate decisions (i.e., whether they take neural or mesodermal routes) are only starting to be precisely defined. Tbx6 expression in the primitive streak region is an early marker of NMP fate decision, as this gene is involved in the induction and specification of mesoderm6,7. Interestingly, early mesoderm cells seem to express high levels of Epha18, and Wnt/β-catenin signalling, as well as Msgn1 were also shown to play important roles in paraxial mesoderm differentiation and somite formation9,10. A complete spatial-temporal analysis of NMPs at a single-cell level will certainly be instrumental to fully understand the molecular mechanisms controlling mesoderm specification.
The formation of somites (vertebrae precursors) is a key feature of vertebrates. During axial elongation, the paraxial mesoderm becomes segmented in a series of bilateral repeating units known as somites. The number of somites and the time required for the formation of new segments varies among species11,12. Somitogenesis involve periodic signaling oscillations (known as the "segmentation clock") that can be observed by the cyclic expression of several genes of the Notch, Wnt and Fgf signalling pathways in the presomitic mesoderm (e.g., Lfng)11,12. The current model of somitogenesis also postulates the existence of a "maturation wavefront", a series of complex signalling gradients involving Fgf, Wnt and retinoic acid signaling that define the position of the posterior border of each new somite. A coordinated interaction between the "segmentation clock" and the "maturation wavefront" is therefore fundamental for the generation of these vertebrae precursor modules as perturbations in these key morphogenetic processes can result in embryonic lethality or in the formation of congenital malformations (e.g., scoliosis)13,14,15.
Despite substantial recent advances in imaging techniques, bioimage analysis methods and software, most studies of axial elongation and somitogenesis still rely on single/isolated two-dimensional image data (e.g., sections), which does not allow a full multidimensional tissue visualization and complicates clear differentiation between pathological malformations (i.e. due to mutations) vs normal morphological variation occurring during embryonic development16. Imaging in 3D has already uncovered novel morphogenetic movements, previously not identified by standard 2D methods17,18,19,20, highlighting the power of in toto imaging to understand the mechanisms of vertebrate somitogenesis and axial extension.
3D and 4D microscopy of mouse embryos, particularly live imaging, are technically challenging and require critical steps during sample preparation, image acquisition and data pre-processing in order to allow accurate and meaningful spatio-temporal analysis. Here, we describe a detailed protocol for live imaging and whole-mount immunofluorescence staining of mouse embryos, that can be used to study both NMPs and mesodermal cells during axial extension and segmentation. In addition, we also describe a protocol for optical projection tomography (OPT) of older embryos and fetuses, that allows 3D in toto visualization and quantification of pathological abnormalities that can result from problems during somitogenesis (e.g., bone fusion and scoliosis)13,21,22. Finally, we illustrate the power of 3D imaging reconstructions in the study and teaching of vertebrate segmentation and axial elongation.
Protocol
Experiments involving animals followed the Portuguese (Portaria 1005/92) and European (Directive 2010/63/EU) legislations concerning housing, husbandry, and welfare. The project was reviewed and approved by the Ethics Committee of 'Instituto Gulbenkian de Ciência' and by the Portuguese National Entity, 'Direcção Geral de Alimentação e Veterinária' (license reference: 014308).
1. Sample preparation for 3D and 4D imaging
NOTE: Here we provide a detailed description on how to dissect and prepare mouse E8.25 to E10.5 embryos for live imaging (1.1), E7.5 to E11.5 embryos for whole mount immunofluorescence microscopy (1.2) and fetuses for optical projection tomography (1.3).
- Sample preparation for live imaging
- Mouse embryo dissection and preparation for live imaging (e.g., LuVeLu reporter 23).
- Dissect mouse embryos between E8.25 and E10.5 in pre-warmed M2 medium (37 °C). Gently remove the yolk sac using clean forceps and wash the embryo once with fresh M2 medium to remove blood and debris produced during dissection.
NOTE: It is important to avoid damaging the embryo in any way, otherwise it will not develop properly during the live imaging procedure. - During the live imaging procedure, incubate embryos in a heated chamber (37 °C), in a 65% O2 and 5% CO2 environment (N2 balanced), in low glucose DMEM medium supplemented with 10% HyClone defined fetal bovine serum, 2 mM L-glutamine and 1% penicillin-streptomycin.
NOTE: These culture conditions allow embryonic development to occur for around 10 h. Other protocols9,10, using different culture conditions, might allow even longer periods.
- Dissect mouse embryos between E8.25 and E10.5 in pre-warmed M2 medium (37 °C). Gently remove the yolk sac using clean forceps and wash the embryo once with fresh M2 medium to remove blood and debris produced during dissection.
- Mouse embryo dissection and preparation for live imaging (e.g., LuVeLu reporter 23).
- Sample preparation for immunofluorescence microscopy
- Mouse embryo dissection and fixation procedure
- Dissect mouse embryos from E7.5 to E11.5 in either cold (4 °C) phosphate buffered saline (PBS) or M2 medium. After removal of all extra-embryonic membranes (e.g., Reichart's membrane or yolk sac) wash the embryo in fresh PBS to remove blood and debris produced during the dissection procedure.
- Fix embryos at 4 °C, in 4% paraformaldehyde (PFA) in PBS, for the time specified in Table 1.
CAUTION – PFA should be handled inside a fume hood
Table 1 – Fixation times for embryos at different developmental stagesDevelopmental stage Recommended fixation time (PFA 4%) E7.5 1h30 E8.5 2h E9.5 3h E10.5 4h E11.5 4h - After fixation, wash embryos at least twice (5-10 min each) in PBS to remove PFA completely. At this point, embryos can be taken straight into the immunofluorescence staining protocol (Step 1.2.2) or can be dehydrated and stored for future use (Step 1.2.1.4).
- If needed, store embryos at -20 °C in 100% methanol for long periods.
- To improve tissue preservation, dehydrate the embryo gradually with 10% increases in methanol concentration (diluted in PBS), each step 10 min at room temperature (RT) on a shaker, until reaching 100% methanol. Replace the 100% methanol once using a fresh aliquot.
- Recover frozen embryos by rehydration following a reverse methanol/PBS series, each step 10 min at RT on a shaker, and with final washes in PBS (twice, 5-10 min each) before entering the immunostaining protocol.
CAUTION – Methanol should be handled inside a fume hood.
- Whole-mount immunofluorescence staining
NOTE: The following immunofluorescence staining protocol was adapted from the procedure described in Osorno et al.24. All washes should be performed on a shaker. After the blocking step, incubations are performed at 4 °C to ensure antibody integrity/preservation.- Wash embryos three times (30 min each) in PBS containing 0.1% Triton X-100 (0.1% PBST) and then once in 0.5% PBST for 1 hour (RT) to improve permeabilization.
- To reduce nonspecific binding, wash in 1 M glycine in PBS (pH 7.5) for 30 min at RT.
- Wash three times in 0.1% PBST for 30 min to completely remove the glycine.
- Incubate the embryos overnight at 4 °C in blocking solution containing: 3% of serum (from the animal species in which the secondary antibodies were produced), 1% of bovine serum albumin (BSA) and 0.1% of Triton X-100, in PBS.
- Dilute primary antibodies in blocking solution (normally 1:200 for T and Sox2 antibodies) and incubate for two to three days at 4 °C.
- Before adding the secondary antibodies, wash embryos three times (30 min each) in 0.1% PBST at 4 °C. Dilute secondary antibodies in blocking solution (1:1000) and incubate for 2 days at 4 °C, protected from light.
- Wash embryos six times (30 min each) in 0.1% PBST at 4 °C.
- For nuclear counterstaining, incubate embryos in 4',6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI), diluted 1:500 in PBST, overnight on a shaker at 4 °C, protected from light.
- Finally, wash embryos three times (30 min each) in 0.1% PBST at 4 °C.
- Tissue clearing and slide preparation
- Tissue clearing using methyl salicylate or a mixture of benzyl alcohol with benzyl benzoate (BABB)
- Before clearing using methyl salicylate or BABB, completely dehydrate embryos by bringing them to 100% methanol, through a methanol/PBS series consisting of successive 10% increases in methanol concentration (incubation times of 10 min each at 4 °C). To achieve complete dehydration, replace the 100% methanol for a fresh one and wait for additional 10 min.
- Perform embryo clearing by embedding in a series of methyl salicylate or BABB in methanol with 20% successive increases in concentration, 20 minute incubation for each step. When the methyl salicylate or BABB solution reaches 100% make two additional changes for a fresh solution.
CAUTION: Both methyl salicylate and BABB should be handled inside a fume hood.
NOTE: For proper clearing, it is essential that embryos are fully dehydrated. It is also essential that the methanol is fully mixed with methyl salicylate or BABB in the clearing solution series. - After the embryos become completely transparent, handle them individually, using preferentially a fluorescence stereoscope, to reduce the chances of tissue damage.
- For imaging, mount embryos in either a 75 mm x 25 mm depression concave glass slide or a 20 mm x 60 mm #1.5 coverglass. The latter option will allow imaging from both sides, if necessary, but it is much more fragile and difficult to seal. Transfer embryos to the microscope slides using a toothpick, a fine cotton tip, a Pasteur pipette or any other similar instrument.
- To avoid compressing the embryos, add spacers made from #1 coverglass shards (170 µm thick), silicone or thin metal washers. Then add a drop of mounting medium, cover with a #1 or #0 20×20 mm coverglass and seal.
- If using glass or metal spacers, seal with melted paraffin to better stabilize the preparation – do not seal with nail-polish because it dissolves with methyl salicylate or BABB. To avoid bubbles, place the coverslip on one side and then, gradually and gently slide it sideways; add more medium if necessary.
NOTE: Please see the video to better understand how to manipulate the cleared embryos and how to mount them on the microscope slide.
- Tissue clearing with RapiClear
- When using RapiClear, embryo dehydration is not required. After Step 1.2.2.9, place embryos in the center of a 75 mm x 25 mm depression concave glass slide, remove all the PBST in the microscope depression slide and add 200 µL of clearing solution.
- After 10 min protected from light, when embryos start to become transparent, replace the clearing solution, wait additional 20 min (protected from light) and then top with a coverslip, starting from one side and gently moving towards the other.
NOTE: The time to fully clear the embryo may go from a few minutes to overnight depending on the stage and size of embryos. Also, the entire process should be done under a stereomicroscope. To better understand the clearing and the microscope slide preparation, please see the video protocol.
- Tissue clearing using methyl salicylate or a mixture of benzyl alcohol with benzyl benzoate (BABB)
- Mouse embryo dissection and fixation procedure
- Sample preparation for optical projection tomography
NOTE: The following procedures were adapted from previous protocols19,25,26. The protocol will be described for the analysis of E18.5 mouse fetuses.- Mouse fetus dissection and fixation procedure
- Dissect E18.5 mouse fetuses in cold (4 °C) PBS, remove all extra-embryonic membranes and then wash the fetuses several times in fresh PBS to remove blood and debris produced during the dissection procedure.
- Fix fetuses in 4% PFA made in PBS, at 4 °C for 5 to 7 days.
- Wash fetuses once (15 min) in PBS and then three times (30 min each) in demineralized water or PBS. Perform the washes on a shaker.
- Dehydrate the fetuses by incubation (25 min series at RT on a shaker) in methanol solutions of increasing concentrations (10% increases diluted in demineralized water or PBS) until 100% methanol.
- Change the 100% methanol solution (use a fresh aliquot) three times (20 min each) to ensure complete dehydration. Embryos can then be stored at -20 °C or be taken directly (one day later) to the bleaching process (Step 1.3.2).
- Bleaching process
- To remove the natural pigmentation, incubate fetuses (separately) on a shaker, first for one day in 5% H202 in methanol and then in 10% H202 in methanol for up to 3 days until the embryos lose all the natural pigmentation.
CAUTION: H2O2 should be handled gently inside a fume hood. The bleaching solution containing H2O2 when in contact with the specimen creates vapors and, therefore, the tube containing the fetuses should remain open during the entire bleaching procedure. Some bleaching protocols suggest the combination of H2O2 with formamide. If this alternative is used, extra-care must be taken, as adding H2O2 to undiluted formamide can result in an explosion. - Gradually rehydrate the embryos in a reverse methanol series (with 10% decrease) in demineralized water, each incubated for 20 min at RT on a shaker, and finally wash three times (30 min each) in demineralized water. Wait one day, change the demineralized water and proceed further.
NOTE: See Supplemental Figure 1 for a representative result of the bleaching process.
- To remove the natural pigmentation, incubate fetuses (separately) on a shaker, first for one day in 5% H202 in methanol and then in 10% H202 in methanol for up to 3 days until the embryos lose all the natural pigmentation.
- Demineralization protocol
NOTE: Demineralization is optional but recommended for fetuses or pups.- Perform demineralization by washing the fetuses in 0.1 M EDTA at 42 °C for a few hours [see also Cho et al.27] followed by five washes (20 min each) with demineralized water to completely remove the EDTA. Perform all procedures on a shaker.
- Sample preparation for clearing
- Embed fetuses in a 1% agarose block. To prepare these blocks fill a 50 mL plastic tube or syringe (building a cylindrical space by cutting off the syringe's exit side and using the plunger to block the opposite side) with melted agarose, place the fetus in the agarose in a vertical position and keep this position at the center of the block with the aid of forceps during solidification of the agarose.
- Place the mold with the block at 4 °C (30 min) for the agarose to fully jellify. In case of incorrect positioning of the fetus within the block or the appearance of air bubbles, melt the agarose by placing the block at 50 °C overnight, and repeat the procedure on the next day.
- Remove the agarose block with the fetus from the mold and place it in a container with demineralized water. Replace with fresh demineralized water after 15 min.
- Before clearing with BABB, dehydrate the specimen. To better preserve tissue integrity, perform fetus dehydration gradually with 10% increases in methanol concentration (diluted in demineralized water or PBS), each step for more than 45 min at RT on a shaker, until reaching 100% methanol.
- Change the 100% methanol (use a fresh aliquot), first four times in one hour intervals and then again on the following day to ensure complete dehydration.
- Clearing process and sample mounting for OPT imaging
- Clear the fetuses on a shaker through a series (2.5 hours each) of BABB solution in methanol, with 25% increases in BABB concentration, until reaching 100% BABB.
- Replace the BABB solution with a fresh one every day, until the fetus is completely transparent. This process might take 3 to 5 days. Keep the block containing the fetus, on a shaker during the entire procedure. Fetuses can stay in 100% BABB solution for long periods.
NOTE: If the fetus is not correctly dehydrated it will become slightly translucent ("whitish" emulsion) after first adding BABB. If this happens, step back into pure methanol and perform two additional washes in fresh methanol. Never refrigerate samples while performing dehydration and clearing, as the changes in temperature may lead to water condensation. - Attach the cleared agarose block to the motor axis of the OPT scanner, adjust optics to obtain an image of the whole fetus and proceed to acquiring a full projection dataset19,28.
- Mouse fetus dissection and fixation procedure
2. Microscope/Image acquisition
- For correct 3D and 4D imaging, choose the microscope that best fits the experimental goal. Table 2 provides general information to guide through the selection.
| Optical microscopy techniques | Imaging principle | Experimental goal and considerations |
| Widefield imaging | Uses fluorescence, reflected or transmitted light. | Ideal for a quick and general overview of the embryo (e.g. for screenings and to assess developmental stages and obvious phenotypes). The reduced depth-of-field, compared to the observable thickness at high magnifications does not allow accurate interpretation or analysis of 3D morphology. |
| Confocal (laser scanning; CLSM) | Uses laser scanning illumination and detection of fluorescence through a pinhole. | Allows imaging of optical slices of fluorescently-labelled samples, ideally with a strong signal. Imaging through a pinhole removes the signal from out of the depth-of-field, thereby allowing accurate discrimination of information in 3D. The acquisition is orders of magnitude slower than widefield, but with unparalleled contrast and 3D discrimination of morphology. High temporal resolution is not achievable because images are acquired 1-pixel at a time. Good for 3D imaging of fixed mouse embryos up to E9.5. Imaging through the whole presomitic mesoderm or somites requires tissue-clearing, because of light-scattering in deeper tissues. Phototoxicity and bleaching is a consideration. Photobleaching can, within limits, be compensated a posteriori. However, phototoxic effects in live samples cannot, and often are not easy to determine. This is more obvious if samples show low expression, and high-laser powers are needed. |
| Two-photon excitation fluorescence (TPEFM) | It uses pulsed near-infrared (NIR) laser excitation instead of visible laser illumination. | TPEFM allows optical slicing through thicker samples than CLSM. Resolution is slightly lower, but the contrast in deeper tissues is considerably better, making it ideal for live imaging of mouse embryos. Although TPEFM is often considered less phototoxic than conventional CLSM, it requires high laser powers which may also have deleterious effects on cells and tissues. Ideal for 3D imaging of embryos up to E11.5, although imaging through the whole presomitic mesoderm still requires tissue-clearing. |
| Confocal (spinning disk) | A form of confocal which, instead of a single laser, uses multiple point-like sources. | The use of multiple point-like structures allows faster creation of optical slices (several frames per second or stacks per minute are achievable) than CLSM. The acquisition is practically as fast as widefield, with reasonable 3D discrimination. However, it only allows imaging of the most superficial tissues of the mouse embryo. Allows more sensitive detection than CLSM, making it an alternative for embryos with low expression of fluorescence proteins. |
| Light-sheet / single plane (LSFM/SPIM) | Instead of widefield or point-like illumination, the sample is illuminated one orthogonal plane at a time. In most configurations, it allows imaging from multiple angles. | Also requires fluorescently-labelled samples. Acquisition of optical slices acquisition is extremely fast (multiple frames per second) and has reduced effects of phototoxicity or bleaching. However, if multiview is required, subsequent dataset pre-processing steps may require hours/days of computation. LSFM/SPIM allows better detection of lower expression levels than CLSM. Ideal for 3D imaging of in toto mouse embryos during gastrulation. Samples often need to be mounted and maintained in suspension (unconventional preparation). |
| Optical projection tomography (OPT) | Optical slices are not detected but calculated from a series of widefield images of the whole embryo from different angles (the “projections”). | Ideal for 3D imaging of later stage mouse embryos/fetuses (>5mm), but only fixed and cleared. Has the advantage of producing 3D stacks of optical slices of both fluorescent and non-fluorescent samples. Datasets are isometric (slices with equal resolution in all three dimensions) making it ideal for anatomical analysis. Acquiring a projection dataset may require only a few minutes, followed by 15-30 min of reconstruction. |
| Optical Coherence Tomography (OCT) | Uses NIR illumination through the sample to obtain optical slices based on interference with light reflection. | OCT allows easy imaging through live tissue (a few millimeters deep into the sample without fluorescent contrast) with a few dozen micrometers resolution. The acquisition is very fast (a few slices per second). Although it is a possible alternative for OPT, this technique is not commonly available. |
| Super-resolution (SR), atomic force (AFM) or near-field imaging (NSOM) | SR is normally based on single-molecule localization and AFM/NSOM on scanning surfaces at sub-diffraction resolutions (few nanometers). | Allows imaging at sub-diffraction level (<200nm resolution), often with the intent to detect single molecules or molecules at the cell surface. Not ideal for morphological analysis of large samples such as mouse embryos. The acquisition is typically a slow process (seconds to minutes per image). |
Table 2 – Generic information to guide the selection of the imaging technique/microscope more suitable for the researcher's specific experimental goal.
3. Image dataset pre-processing
NOTE: Here we highlight some of the key steps of image dataset pre-processing, namely noise reduction (3.1) and deconvolution (3.2), and provide algorithms that allow proper preparation and pre-processing of 3D datasets time-series (3.3) and whole-mount immunofluorescence stainings (3.4). Finally, we indicate references that describe in detail a protocol for OPT dataset pre-processing and reconstruction.
- Noise reduction
- If the images show reduced signal-to-noise ratio, consider applying a classical method such as a "Median" filter or "Anisotropic diffusion" available in Fiji/ImageJ29, or a more elaborate method based on Machine Learning such as "CARE"30 or "Noise2Void"31.
NOTE: This is especially relevant for live imaging datasets, where the photobleaching and photodamage is a major concern, and lower exposures and higher detector gains are necessary.
- If the images show reduced signal-to-noise ratio, consider applying a classical method such as a "Median" filter or "Anisotropic diffusion" available in Fiji/ImageJ29, or a more elaborate method based on Machine Learning such as "CARE"30 or "Noise2Void"31.
- Deconvolution
- If the images were acquired with high-resolution (near or at Nyquist sampling) consider performing image restoration with deconvolution to enhance the quality of the dataset before analysis. Beware that some deconvolution tools also include a denoising step.
NOTE: This has been extensively addressed in Krull et al.32 and in documentation available at the Huygens deconvolution software website (https://svi.nl/HomePage).
- If the images were acquired with high-resolution (near or at Nyquist sampling) consider performing image restoration with deconvolution to enhance the quality of the dataset before analysis. Beware that some deconvolution tools also include a denoising step.
- Preparing a 3D dataset time-series for proper visualization and analysis
NOTE: Often embryo or stage drift can occur during time-lapse imaging. This needs to be corrected before performing analysis. Sometimes, the drift is severe enough that the acquisition needs to be interrupted and adjustments made. In this case, it is best to keep the same X, Y and Z dimensions.- Separate 4D stacks can be concatenated into a single 4D hyperstack using Fiji's "concatenate" function. However, this tool does not handle composite/hyperstacks, so it is necessary to convert all 4D segments into simple stacks using the operation Image | Hyperstacks | Hyperstack to stack. Keep some numbering system to maintain the order of these 4D segments and take note of the information about each one (channels, slices, time-points). Repeat the procedure for all segments and then concatenate them using the procedure Image | Stacks | Tools | Concatenate.
- Reconstruct the concatenated hyperstack with the operation Image | Hyperstacks | Stack to Hyperstack and choose the correct order of the different dimensions. Inspect the Hyperstack to make sure all dimensions are correctly sequenced.
NOTE: The number of Channels (c) and Slices (z) should be the same for all 4D segments; the number of (time) Frames (t) should be equal to the total of time points added for all 4D segments. It is important to browse through the stack to understand how the different dimensions are interpolated, before performing the conversion to hyperstack. Fiji/ImageJ default is alternating channels, then alternating Z slices, and then alternating time-points ("XYCZT"). Confirm that this applies to the data generated by the microscope. - Choose Composite as the Display mode and save as Tagged Image File Format (TIFF). For large datasets, consider converting to BigDataViewer33 format, by performing the operation Plugins | BigDataViewer | Export Current Image as XML/HDF5. This generates a dataset that can be browsed more efficiently and in 3D using the BigDataViewer and can be handled by other Fiji/ImageJ tools for big-data datasets.
- Register the concatenated hyperstack (time-series of 3D stacks) to compensate for embryo/stage drift using either the "Correct 3D Drift" ImageJ plugin34 or the BigStitcher plugin35, depending on the degree of the drift correction needed. XML/HDF5 files can be opened with BigStitcher.
NOTE: It is necessary to perform the registration of 3D time-lapses that show embryo drift before any attempt to perform cell tracking or movement analysis; correcting for drift is also necessary to properly interpret morphogenetic movements.
- Pre-processing of datasets of immunofluorescence imaging
- Z-depth signal attenuation
NOTE: Although the method we propose to correct signal attenuation can be useful, this should be used carefully, as often less signal in depth may actually reflect a biological and not an optical phenomenon. Consider measuring the decay in an area of tissue where the molecule of interest is not expected to be present or expressed and adjust the compensation for that area. If there is still a lower positive signal in depth, it may reveal an actual biological phenomenon. Consider also that some areas of the sample may produce more scattering or laser attenuation than others, and that it will not be fully corrected with this simple method.- For thicker/late-stage embryos, beam attenuation, photobleaching and spherical aberration caused by mismatch of refractive indices (between the mounting medium and the microscope objective) can result in datasets where the fluorescence intensity is greatly attenuated in the deeper slices of the Z-stack. Therefore, compensate for this loss of signal before analysis. Determining analytically the most accurate attenuation is complex and highly sample dependent36, but a quick approximation can be determined empirically in ImageJ/Fiji using the Process | Math | Macro function and clicking on Visualizar.
- Load the Z-stack in Fiji/ImageJ and then perform the procedure Image | Stacks | Reslice. Start at: top, keep Avoid interpolation ticked and OK. Now the "Z" will be the "Y" axis. Next, do Image | Lookup tables (LUT) | Fire (or any other multi-colored LUT). This will help to assess visually the best compensation during the next steps. Switch to a slice in the middle of the embryo, where the effects of attenuation in depth are clearly visible (the intensity drops from the top [superficial] to the bottom [deeper]).
- Proceed to determining the correction of the vertical ("y") intensity drop with the procedure Process | Math | Macro, and add the following "Code" exactly as written here:
v = v * A * exp ( B * y/h )
In this expression, "v" is the variable for pixel intensity (which will be adjusted as a function on depth), "y" the variable for depth, and "h" for full depth, and "exp" is an exponential function; "A" should be replaced with a number between 0.5 and 1.0 (choose lower values to avoid oversaturation of the top layers of the Z-stack); B replaced with a number between 0.5 and 2.0, depending on how severe the Z-depth attenuation is – ideally it should be 1, however in some cases less (or more) is necessary to correctly compensate the bottom layers; a higher value of B will result in more attenuation compensation. Click on Visualizar to make a first assessment. Test different values of A and B until obtaining adequate compensation from top to bottom of the image (the "Fire" LUT can be helpful for this assessment). When satisfied, apply the settings by clicking OK.
NOTE: This must be performed in each channel individually, because red-shifted dyes and lasers may attenuate less, and some dyes bleach faster than others. Keep in mind that this procedure might not be accurate enough to allow reliable quantification of fluorescence intensity variations in depth, although it is certainly more reliable than performing quantifications directly in the original 3D dataset that is severely affected by attenuation of signal in depth. One way to identify and control for this effect, is to compare imaging different embryos from the dorsal or ventral sides. - Restore the original geometry of the compensated Z-stack by doing Image | Stacks | Reslice, start at top, keep Avoid interpolation ticked, and now the "Y" must become the "Z" plane again; replace the color by performing "Image | Lookup tables | Grays" (or the other LUT of choice). Discard the original non-compensated z-stack and save the compensated version.
NOTE: See Supplemental Figure 2 as a representative result of the Z-depth signal attenuation method.
- Z-axis scaling correction
- If imaging with an objective designed for a refractive index different to that used for mounting the embryos, it is necessary to perform re-scaling of the slice thickness, otherwise measurements in depth will be incorrect (potentially by 50% if using a "dry" objective on a tissue-cleared embryo). This is explained, reviewed and the different methods discussed, in 37 and 38. Determining analytically the actual Z-axis distortion scale is complex, but an acceptable approximation can be easily determined by finding the ratio between the refractive index of the sample and the refractive index of the objective (e.g., 1.53/1.0 for a methyl salicylate-cleared embryo imaged with a dry 20 x objective, or 1.56/1.33 for an embryo cleared with BABB and imaged with a water immersion objective).
- With the Z-stack dataset already opened in Fiji/ImageJ (for multichannel images, after they were assembled as "composite" with all channels), go to Image | Properties and change the Voxel depth to the slice thickness obtained during image acquisition multiplied by the Z-axis scaling correction determined in the previous step (4.2.1; "Image dataset pre-processing" section). Confirm the result using the Image | Stacks | Orthogonal Views.
- Repositioning of embryo to an anatomically standard position using Fiji/ImageJ
NOTE: Repositioning the embryo (see the advantages highlighted in Supplemental Figure 3) into a standardized anterior-posterior [A-P] and dorsal-ventral [D-V] axis position using Fiji/ImageJ, requires the installation of the TransformJ 39 suite of plugins from Fiji's "ImageScience" update website.
NOTE: This technique is preferable to rounds of rotations in the three planes which would introduce aliasing artifacts and degradation of resolution. However, because the Fiji/ImageJ 3D viewer plugin cannot handle properly datasets larger than 200-300 Mb (the plugin may not render or show unpredictable behavior when trying to rotate the viewing angle), the original 3D dataset must be down-sampled first.- Start by reducing the dataset size in Fiji's Image | Scale and then insert the X, Y and Z "scale" values necessary to reduce the dataset to less than 200 Mb (for example a 0.5 x 0.5 x 0.5 down-sampling will result in a dataset 8 times smaller). Make sure the Create new window option is ticked.
- Then go to Plugins | 3D viewer. Inside the 3D viewer window, click over the embryo to select it, and a red 3D box should appear. When using this mode (with the red box activated), the rotation of the dataset can be controlled with the mouse, in contrast to the default behavior, which is to rotate the viewing angle. When repositioning the embryo with the mouse, never click outside or the dataset will be deselected.
- When satisfied with the new position of the embryo, select Edit | Transformation | Export transformed image in the menu of the "3D viewer" window. To inspect the different orthogonal planes go to Image | Stacks | Orthogonal Views. If the embryo is not correctly positioned, close the window and go back to the 3D viewer window and re-adjust. Repeat step 4.3.2.
- When the embryo is correctly positioned, select from the "3D viewer" window menu Edit | Transformation | Save transform and save the transformation matrix to a text file with the *.mat extension. Open this text file using Fiji/ImageJ, remove the first two lines (text) and re-save. This creates a transformation matrix file compatible with the "TransformJ" plugin described in 39.
- Switch to the full-resolution dataset (can be a multi-channel composite hyperstack) and perform the operation Plugins | TransformJ | TransformJ affine. In the new window, browse to search for the matrix file previously saved, select the Cubic B-Spline interpolation | resample isotropically | OK.
NOTE: This operation is memory and CPU intensive. Depending on the workstation and dataset size, the operation may take from minutes to hours. The use of the resample isotropically option guarantees that no resolution is lost during the transformation operation, so the dataset will increase in size significantly and will no longer be anisotropic like the original confocal z-stack; it is to be expected that the number of slices in the Z-axis increases to the same number of X and Y pixels of each slice, and the stack bloated to 5-10x the original size. This is necessary to guarantee that no information is lost in the course of interpolation of voxels during rotation and translation. - Once the embryo is properly repositioned it is often possible to trim most of the empty space created around the embryo. Perform a full Z-projection Image | Stacks | Z Project… and choose Maximum intensity; draw the minimum ROI that contains the whole embryo in X and Y. Switch to the original dataset image windows, go to Edit | Selection | Restore selection, and crop by selecting Image | crop.
- Trim the slices in the beginning and end that do not intersect embryo tissues. For this, select Image | Stacks | Tools | Slice remover and specify the first and last slice to remove, making sure to change the "increment" to 1 (otherwise it removes only every other slice).
- After repositioning and trimming the new dataset make sure to save as a new TIFF file. If this dataset is too large (more than the GPU RAM) consider using BigDataViewer to export as XML/DHF5, as explained in step 3.3 (from the "Image dataset pre-processing" section).
- Z-depth signal attenuation
- OPT dataset pre-processing and reconstruction
- Use the protocol for OPT dataset pre-processing and reconstruction described in Martins et al.19 and Gualda et al.28.
4. 3D rendering, visualization and analysis
NOTE: Here we provide a list of possible applications of different software tools, that allow or enhance the visualization and analysis of 3D imaging datasets.
- 3D rendering and visualization using Drishti
NOTE: Drishti40 is a free scientific visualization software designed to explore and present 3D and 4D datasets from micro-CT, confocal/multiphoton and light-sheet microscopy. Here we use this software for 3D rendering and visualization of whole-mount immunofluorescence staining (Step 2; "Sample preparation for 3D imaging" section). There are multiple tutorials online to help users understand how to operate Drishti; search online for "Ajay Limaye Drishti 3D tutorials". Drishti cannot directly read stacks of TIFF files, so they must be converted first to the native "pvl.nc" format.- Run the "drishtiimport.exe" tool, located in the Drishti installation folder: Files | Load | Files | choose "Grayscale TIFF image files, and load a single-channel 3D stack, as saved from Fiji/ImageJ. In the case of a multi-channel composite stack, split the channels in Fiji/ImageJ and save each individually. Voxel type is "ushort"; "File"…"Save as"…"TIF", reply "y" to all questions. In the Additional information window asking for voxel size make sure to add the correct "X Y Z" voxel sizes.
- Loading *.pvl.nc files into Drishti and rendering: In the main window of Drishti, File | Load | Load 1 ( – 4 ) volumes depending on the number of channels available (as separate files). After loading, press F2 for high-quality render. This action requires a powerful GPU card. Information to handle the rendering parameters, Transfer function editor, search on the online tutorials. Once satisfied with the rendering properties proceed to generating an animated rendering.
- Go to View and activate the Keyframe editor. Handle the embryo and position it in the desired starting position. Use the right-mouse button to "drag" the embryo to the center if necessary, or the mouse scroll to zoom. Hit set keyframe de Keyframe editor, then choose another position, angle or zoom, drag the time-line marker to a different time point and Set keyframe again. Now there are 2 keyframes where the embryo is represented in 2 different positions/angles/zoom, and a number of frames in between. Pressing the Play button, Drishti can interpolate the missing conditions and play it as an animation. Go to File | Save image sequence to save the animated sequence of all frames. This frame sequence can later be opened in Fiji/ImageJ and saved as AVI (File | Import | Image sequence … File | Save As | JPEG compression with a reasonable frame rate (we suggest 15 – 30).
NOTE: Although Drishti allows the saving of *.wmv videos, it is advisable to instead save as separate frames and only later convert the series to AVI or MOV video format (this can be done using Fiji/ImageJ).
- 3D reconstitutions and manual segmentation of tissues using Amira
NOTE: This commercial software allows, manual or automatically/semi-automatically, 3D segmentation of embryonic tissues in 3D datasets. There is an online "Learning Center" for this software with multiple tutorials available 41. Below are described the basic steps required to manually segment embryonic tissues from immunofluorescence-stained embryos (step 1.2). Manual segmentation is much easier after the embryo is correctly positioned in a standard A-P/D-V axis (see the" Image dataset preprocessing" section, step 4.3).- Load a 3D TIFF dataset. Make sure to specify the correct voxel dimensions, when asked. Create and connect a visualization module (e.g, "Orthoslice" or "Volren") and inspect the dataset in 3D.
- Connect the Label field (or Edit New Label Field in newer versions of Amira). In this software, results of manual segmentation are stored in "materials", so create one material for each tissue of interest. Use the "lasso" tool for manual segmentation. There are several tutorials available explaining how to perform this (search online for "Amira Segmentation Editor tutorial").
- When finished with segmentation of all tissues of interest, exit the Segmentation Editor by switching back to Project mode. Having created a new version of the dataset (the "labels field", which is now attached to the original dataset module) in which pixels belonging to each material have the same value.
- Convert these "materials" into 3D objects by generating 3D surface models from the "Labels" dataset. This can be done by attaching a "Generate Surface" module, adjusting parameters as necessary (activate the "compactify", "border", "Adjust coords" and "Unconstrained Smoothing" options). Click Apply and a new module *.surf is generated.
- Visualizing the surface objects, extracting and exporting individually as *obj files. Attach a Display | SurfaceView module to the *.surf module, and inspect in the main viewer. In the "properties" panel of the "SurfaceView" module, in the "Materials" property select Todos + Todos and click remove, so the viewer's buffer is emptied. Then in the second pull down of the Materials property select only one material and click Add to the buffer; only that material should be visible.
- In the property Draw style click in more options | create surface and a new *.surf module is created and added to the project. Rename it to the name of the tissue (press F2 on the keyboard), and File | Export Data As… and Save as type: Wavefront (*.obj). Repeat the operation for each material.
NOTE: These *.obj files, each containing one of the tissues segmented in Amira, can be used by other tools ("3Dviewer" plugin from Fiji/ImageJ and SimLab).
- Interactive 3D visualization within in a portable document format (PDF) using SimLab Composer
NOTE: This 3D Computer Graphics software facilitates the creation of surface rendered images, animations and simulations. Useful tutorials can be found on line 42. Here we describe how this software enables the creation of 3D PDF interactive illustrations, using the wavefront (*.obj) 3D surface files created with Amira (Step 2.3; "3D rendering, visualization and analysis" section).- Go to File | New and create an empty scene. Then File | import and import the 1st *.obj file saved from Amira. Keep all options of the Import file window unticked and click OK.
- If nothing appears in the Composer's display window, click Ctrl+F or the icon to Fit all. Now the segmented object should appear in the center of the window. Repeat the process to add another segmented object and confirm its position relative to the 1st one (for example, 2 adjacent somites).
- Select one of the objects in the display window by clicking with the left mouse button, change its name to reflect the name of the anatomical structure or tissue, then switch to the "3DGeom~1" representation, and using the Materials panel, change the color by altering the R,G,B values.
- Repeat for all objects, until the 3D illustration is fully assembled. Note that some materials can also be made transparent by manipulating the "Alpha" channel, next to the RGB values, and thereby allow observation of internal or occluded objects.
- With this software the assembled embryo illustration can be exported as a "3D PDF" file, which can be included in publications. First create a "template" with text and active links to change "Scene states" to be able to include instructions and three different stages in the same illustration. This can be done by switching from "Scene building" to "Sharing" mode (buttons on the left of the Composer window), then clicking in Show PDF settings, and creating a new page template. To create a simple interactive figure to include in a manuscript, do not use a template and simply Export PDF.
NOTE: Use Adobe Acrobat Reader software to interact with the 3D PDFs (operability of the functions in the interactive 3D PDF illustration may vary depending on versions of Acrobat Reader available). Most other viewers are not compatible with the 3D PDF format, including web browsers. Such 3D PDF interactive illustrations can be included in publications; ask editors about this possibility in advance.
- 3D visualization and analysis using Imaris
NOTE: This software allows comprehensive imaging visualization, analysis and interpretation of 2D-4D microscopy datasets, with intuitive interface and workflows. There are several useful tutorials online on how to get started with this software, available at the website (https://imaris.oxinst.com/tutorials). To load and interact more effectively with large datasets in Imaris (typically, those larger than the GPU memory) it is advisable to convert the dataset first to “*.ims”, using the “ImarisFileConverter.exe” program installed in the Imaris installation folder. The ImarisFileConverter can read many microscopy image formats, including OME and “h5” files as saved by Fiji’s BigDataViewer.- 3D visualization
- This software can render a 3D visualization of the dataset using the "3D View" mode and the user is able to make several adjustments to how the dataset is displayed [e.g., choose between MIP (Maximum intensity Projection) or blend mode (better rendering with depth cues and shadows)].
- Orthogonal views" mode – With proper embryo positioning (Step 4.3; "Image dataset pre-processing" section) one can use the "Section" tab and navigate through the entire embryo and see optical sections from the normal planes (transverse, coronal and sagittal). If embryos are not properly repositioned, interpreting their anatomy with orthogonal planes is extremely difficult (see Supplemental Figure 3). Although Imaris has a function known as "reference frame" to work around this problem when analyzing 3D embryos, it is preferable to reposition the datasets a priori.
- 3D analysis
NOTE: This software also has several tools for analyzing morphology and fluorescence intensities. As an example, here we describe a simple workflow to analyse tissue fluorescence intensity variations over time using the Imaris "Spots" tool, where the user manually places spherical regions of interest (ROIs) in the embryo in 3D, and Imaris can then measure the tissue fluorescence inside these spherical ROIs.- Load a 3D or 4D dataset in Imaris and add new spot in the 3D view mode. Then, one can select specific points of the embryo (also during several time points if one uses a 4D dataset) and quantify inside those spots, for example, the intensity mean.
NOTE: This software also allows plotting the intensity over time. This allows the user to easily answer questions like: "how does the fluorescence change inside a somite over time?". - Add the spot module by clicking on the Add new spot button or selecting 3D view | Spots in Imaris menu. Choose Skip automatic reaction, edit manually. Switch the Imaris pointer mode from Navigate para Select (this can also be done by hitting the ESC key on the keyboard).
- On the Spots properties window, select the Specific channel to which the spot will be attached (e.g., "Channel 1"), and activate in Manual tracking the Auto-connect to selected spot and the Enable delay before auto-advancing options.
- Move the mouse cursor to the visualization window and the cursor should change to a hollow yellow wire sphere. Go to time-point 1 and add a spot (=sphere) by clicking in the tissue of interest. The sphere ("Spot") is only added if clicking while holding the shift key. Repeat for all the desired time points making sure to track the same portion of tissue over time.
- In the Spots Properties window, switch to the Estatística tab, and inside the Estatística switch from Overall para Detailed statistics. From the first pulldown choose Specific values, and from the second pulldown choose "Intensity mean Ch=*…", where Ch* is the channel in which the measurement will be performed. A button right to the "Click to open time-plot panel" will be found in the bottom left of the spots properties window. Clicking this button opens up a plot with the median fluorescence intensity inside the "spots", plotted over time. These values can also be exported to a spreadsheet for further analysis.
- Load a 3D or 4D dataset in Imaris and add new spot in the 3D view mode. Then, one can select specific points of the embryo (also during several time points if one uses a 4D dataset) and quantify inside those spots, for example, the intensity mean.
- 3D visualization
Representative Results
The representative results shown in this paper for both the live and the immunofluorescence imaging, were obtained using a two-photon system, with a 20 × 1.0 NA water objective, the excitation laser tuned to 960 nm, and GaAsP photodetectors (as described in Dias et al. (2020)43. Optical projection tomography was done using a custom built OPenT scanner (as described in Gualda et al. (2013)28.
Live imaging (4D analysis)
A representative analysis of LuVeLu reporter activity in mouse embryos during axial extension, obtained according the described protocols for "Sample preparation for live imaging" (Step 1.1; "Sample preparation for 3D imaging" section), "Live imaging dataset pre-processing" (Step 3; "Image dataset pre-processing" section) and "3D visualization and analysis using Imaris" (Step 4.4; "3D rendering, visualization and analysis") can be observed in Figure 1 and Video 1.
3D visualization and analysis
A representative result for the use of immunofluorescence assays to detect potential NMPs and key regulators of mesoderm differentiation, obtained following the protocols described for "Sample preparation for immunofluorescence microscopy" ("Sample preparation for 3D and 4D imaging" section, step 1.2), "Deconvolution" and "Immunofluorescence imaging dataset pre-processing" ("Image dataset preprocessing" section, step 3.2 and 3.4 respectively), and "3D visualization and analysis using Imaris" ("3D rendering, visualization and analysis", step 4.4) can be observed in Figure 2.
3D renderization of immunofluorescence stainings
Video 2 shows a representative result for an immunofluorescence assay to detect potential NMPs obtained according to the described protocols for "Sample preparation for immunofluorescence microscopy" (Step 1.2; "Sample preparation for 3D and 4D imaging" section), the "Deconvolution" and "Immunofluorescence imaging dataset pre-processing" (Step 3.2 and 3.4, respectively; "Image dataset pre-processing" section), and the "3D rendering and visualization using Drishti" (Step 4.1; "3D rendering, visualization and analysis" section).
3D reconstructions
Video 3 was generated following the described methods for "Sample preparation for immunofluorescence microscopy" (Step 1.2; "Sample preparation for 3D and 4D imaging" section), "Immunofluorescence imaging dataset pre-processing" (Step 3.4; "Image dataset pre-processing" section), and "3D reconstructions and manual segmentation of tissues using Amira" (Step 4.2; "3D rendering, visualization and analysis"). It shows a 3D reconstruction, based on immunofluorescence staining, of the caudal tissues of a E9.5 wild type (WT) mouse embryo.
Interactive 3D illustrations
In Figure 3 we present an interactive 3D visualization of a reconstruction of the caudal tissues of mouse embryos in a portable document format (3D PDF), prepared following the described protocols for "Sample preparation for immunofluorescence microscopy" (Step 1.2; "Sample preparation for 3D and 4D imaging" section), "Immunofluorescence imaging dataset pre-processing" (Step 3.4; "Image dataset preprocessing" section), "3D reconstructions and manual segmentation of tissues using Amira", and "Interactive 3D visualization within in a portable document format (PDF) using SimLab" ("3D rendering, visualization and analysis", Step 4.2 and 4.3 respectively). This type of format can be easily used to facilitate communicating scientific contents to a more general audience, particularly in a teaching environment. Interactive visualization requires the use of Adobe Acrobat Reader (allow the 3D plugin).
OPT dataset visualization
Video 4 shows a representative example of an OPT reconstructed dataset, after imaging a E18.5 mouse fetus. Sample was prepared as described in protocols for "Sample preparation for optical projection tomography" ("Sample preparation for 3D and 4D imaging" section; Step 1.3) the "OPT dataset pre-processing and reconstruction" (Step 3.5) and different modules of the "3D rendering, visualization and analysis" (Step 4). A detailed anatomical analysis (e.g., measurements) can also be performed in this dataset using Fiji/ImageJ, Amira or the Imaris software. The movie contains four video segments: the first shows a sequence of black and white sagittal slices produced with Fiji/ImageJ; the second, orthogonal views produced with Imaris showing interactive 3D slicing and rendering of orthogonal tissue portions; the third shows the fetus "assembling" from colored sagittal slices, as prepared in Amira; the last video segment display an animation of the fetus in 3D from different views, prepared with Drishti.
Video 1 –Two-photon live imaging of LuVeLu reporter expression in Snai1-cKO and control E8.5 embryos (adapted from ref. 43). The first segments show the reporter activity at time = 0 (z-stack of 5 µm step-size) and the second parts contains the full live imaging (10 µm z-stacks every 8.5 min). Please click here to download this Video.
Video 2 – 3D renderization of a whole-mount immunofluorescence staining for T (Brachyury) and Sox2 of a E10.5 wild type tailbud. T expression is shown in magenta, Sox2 in yellow and DAPI in grey. Please click here to download this Video.
Video 3 – 3D reconstruction of the caudal part of a late E9.5 wild type mouse embryo, with a focus on the contouring of different caudal tissues including mesoderm. Please click here to download this Video.
Video 4 – E18.5 wild type embryo as imaged with optical projection tomography, highlighting the in toto imaging and several techniques for visualizing and rendering 3D datasets. The first segment shows a sequence of sagittal slices prepared in Fiji/ImageJ, the second "live" orthogonal views as rendered in Imaris, the third a sequential rendering from colored sagittal slices prepared in Amira, and the fourth an animated rendering prepared in Drishti. Please click here to download this Video.
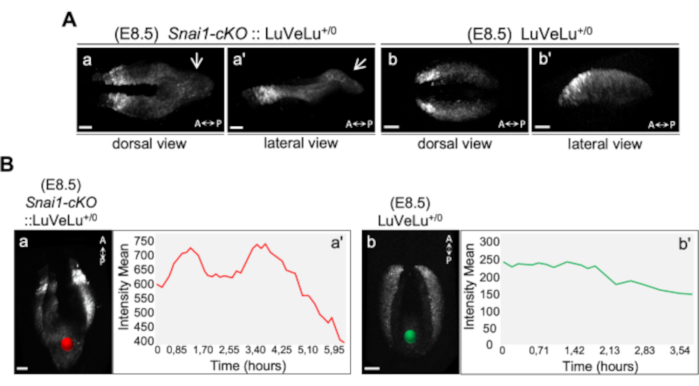
Figure 1: 4D analysis of LuVeLu reporter expression in E8.5 Snai1-cKO and control embryos (adapted from ref. 43). (A) Snapshot at time-point = 0 of the LuVeLu reporter, in Snai1-cKO(Meox2-Cre+/0::Snai1flox/-) (Aa) and control (Ab) embryos. In addition to the normal LuVeLu signal in the presomitic mesoderm, Snai1-cKO embryos also display LuVeLu expression in the ectopic bulge that arises from the primitive streak (white arrows). (B) Snai1-cKO::LuVeLu+/0 temporal intensity mean quantitative analysis in the region highlighted by the red spot (Ba) indicates the existence of two-peaks (at t = 1.3 h and t = 3.6 h of the time-lapse) and a substantial decrease between them, therefore suggesting cycling activity in the bulge of the conditional mutant embryo. No signs of LuVeLu cycling activity were observed near the primitive streak (green spot; Bb) in LuVeLu+/0 control embryos. Scale bar: 50 µm. Please click here to view a larger version of this figure.
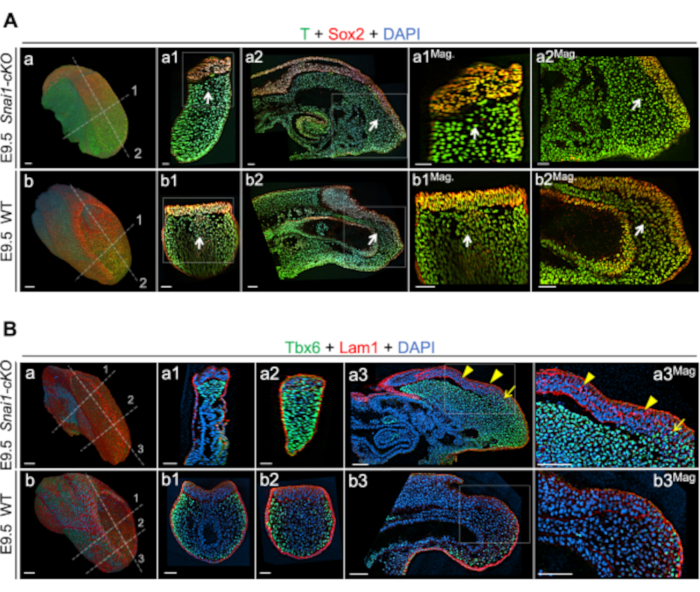
Figure 2: Whole-mount immunohistochemistry in E9.5 Snai1-cKO and WT embryos (adapted from ref. 43). (A) Immunostainings for T (green) and Sox2 (red). (B) Immunostainings for Tbx6 (green) and Lam1 (red). DAPI is shown in blue. 3D rendering (blend mode) of the caudal part of the different embryos (Aa, Ab, Ba, and Bb). Transversal (Aa1, Ab1, Ba1, Ba2, Bb1 and Bb2) and sagittal (Aa2, Ab2, Ba3 and Bb3) optical sections together with magnifications (Mag.) are also shown for the different embryos. Magnifications are shown without DAPI. White arrows highlight differences in the location of some NMPs (T and Sox2 double-positive cells) in the mutant embryos. Yellow arrows and arrowheads point to ectopic/abnormal Tbx6 and Lam1 expression respectively, in the Snai1-cKO embryos. Scale bars correspond to 50 µm. Please click here to view a larger version of this figure.
Figure 3 – 3D PDF – Interactive 3D illustration in 3D PDF format showing the 3D reconstructions of segmented caudal tissues of wildtype mouse embryos during axial extension (E8.5, E9.5 and E10.5). Segmentation was done manually in Amira, and the 3D PDF was assembled and exported in Simlab Composer. Interactive visualization requires Adobe Acrobat Reader (allow the 3D plugin). Please click here to download this figure.
Supplemental Figure 1 – Representative result of a bleached embryo (after Step 3.2; "Sample preparation for 3D and 4D imaging" section). Please click here to download this File.
Supplemental Figure 2 – Representative result of Z-depth signal attenuation (Step 4.1; "Image dataset pre-processing" section). A – before Z-depth signal attenuation; B – good Z-depth signal attenuation; C – incorrect Z-depth signal attenuation. Please click here to download this File.
Supplemental Figure 3 – Representative result of a whole-mount immunofluorescence staining showing and E7.5 mouse embryo that has been repositioned (A) following “Repositioning of embryo to an anatomically standard position using Fiji/ImageJ” (Step 3.4.3; "Image dataset pre-processing" section) versus the same embryo without being repositioned (B). Visualization was obtained using Imaris (see Step 4.1.2; "3D rendering, visualization and analysis" section). T in yellow, Sox2 in magenta and DAPI in blue. Please click here to download this File.
Discussion
Axial elongation and segmentation are two of the most complex and dynamic processes occurring during vertebrate embryonic development. The use of 3D and 4D imaging with single-cell tracking has been applied, for some time, to study these processes in both zebrafish and chicken embryos, for which accessibility and culture conditions facilitate complex imaging19,44,45,46,47,48,49. In contrast, the mid and late organogenesis stages of the mouse embryo remain poorly studied in such detail, although some advances have been made, for example in intravital imaging50. In this manuscript we provide specific protocols for several methodologies that can be used for the acquisition of multidimensional images and their analysis to facilitate the study of NMPs and their mesoderm derivatives during neck, trunk and tail formation. Although these protocols were designed for mouse embryos, they can be easily tuned to work for explant systems and in vitro models such as 3D embryonic stem cell aggregates like gastruloids51,52. Indeed, if applied to human gastruloids53 these methods would facilitate a better in toto imaging and data analysis of human axial elongation and segmentation, especially given the compatibility of these in vitro model systems with long-term 4D live imaging. In addition to these protocols, we also provide general information about different available microscopy techniques and how they can be used to fit specific experimental goals. We hope this information helps researchers to improve their experimental designs and to take full advantage of the microscopy equipment and image analysis tools available in their laboratories and institutions.
Sample preparation is one of the most important steps of the protocols described in this manuscript, as proper preservation and processing of the embryo during the whole procedure, from dissection to mounting in the microscope, is essential to obtain high quality data. Dehydration (and rehydration), bleaching and clearing procedures are the most critical steps during sample preparation, strongly influencing the final outcome of the imaging. Therefore, we have detailed these steps in our protocol and provided tips that will help researchers to achieve a spotless preparation of their samples. Particularly, we have detailed the use of three different solutions for clearing post-implantation mouse embryos (up to E11.5). Although methyl salicylate and BABB have been used for several years and are efficient clearing reagents, complete laser penetration in some tissues, particularly mesoderm, is sometimes difficult to achieve. Conversely, RapiClear was found to be very effective in clearing mouse embryos at these developmental stages. In our hands, it proved to enable complete laser penetrance in the various embryonic tissues and, since it is not toxic and does not require embryo dehydration, it greatly simplifies the key step of mounting the embryos in the microscopy slide. Also, to overcome the necessity of manipulating the embryos in the slide (cleared embryos become very fragile and difficult to manipulate once they are transparent) or the need to resort to commercial software (e.g., Amira or Imaris), we have provided a simple but efficient pipeline, using Fiji/ImageJ (free open-source software), that allows embryo repositioning to an anatomically standard/specific position during image pre-processing without losing data quality. Therefore, we expect the methods and details that we provide in this manuscript to facilitate and contribute to improve the key step of sample preparation during immunofluorescence assays.
During image acquisition, it is important to consider the configuration of the system, which affects how samples need to be maintained for live imaging or mounted for optimal observation in 3D. For example, mouse embryos require a heating source, high levels of O2 and liquid nutrient-rich media (not solid), making it more difficult to be kept live in a microscope with an upright, or a conventional light-sheet configuration. For correct 3D imaging of embryos, immobilization is also important. Available optics is also another important factor to consider; for proper 3D imaging, high numerical aperture objectives should be preferred however those are not always available, especially when large working distances are required (more than a few hundred micrometers). Physiological (i.e., "dipping") lenses are very common for live imaging however these are not ideal for imaging through a glass-bottom dish or for cleared samples mounted on BABB or methyl salicylate. Although objectives optimized for cleared tissues are becoming more common, they are still relatively rare to find in most laboratories. It is also possible to image cleared tissues with conventional objectives however users must be aware of the limitations and the necessary care before interpreting and analyzing the datasets, as we explain in the sections above. Furthermore, accurate detection and interpretation of imaging results always require careful choice not only of the instrument but also of the operating conditions and setting of parameters. Users are encouraged to read the following reviews 54,55,56,57 to fully understand the principles of proper digital imaging and interpretation of bioimages.
Immunofluorescence applied to (2D) histological sections has been the hallmark of studies seeking to understand tissue and cell organization inside embryos. The emergence of whole-mount immunolabeling methods and non-destructive 3D imaging techniques (as those described here) has provided developmental biologists with the means to investigate tissue and cell spatial organization in intact embryos, where it is easier and more accurate to understand gene and protein expression and their relationship to morphogenetic processes. With in toto imaging, it is now possible to perform virtual histology (virtual sectioning in arbitrary slices; Supplemental Figure 3), and the 3D visualization and analysis tools can be used to easily explore different hypotheses about cell and tissue architecture and communication (see Figure 2, Video 2 and Video 4). Considering that many journals (e.g., eLife and Development) are now giving the opportunity to include videos in the online version of the paper, we urge researchers to take advantage of this opportunity and not only perform 3D and 4D imaging experiments and analysis but also to make 3D videos highlighting their results. This important change in the way data is presented and published, will enable a better comprehension of the results by the researchers and their peers. Ultimately, these methods have the potential to improve our understanding of vertebrate segmentation, particularly regarding the role of NMPs (Attardi et al. (2018)45 and Dias et al. (2020)43 are two good examples).
In this work we have highlighted how the use of some software tools (e.g., Amira and Imaris, but also free open-source tools like Fiji/ImageJ and Drishti) allows and enhances 3D data visualization and analysis in the context of vertebrate axial extension and somitogenesis. In most cases, like for several bioinformatic tools, the software described here can potentially be replaced by another (i.e., there are several software solutions and redundancies). However, we find that the "3D View" function in Imaris allows a 3D visualization with much better quality than the one obtained using the "3D Viewer" Fiji/ImageJ plugin, allowing for example to choose between MIP or blend modes. In addition, we find the "Orthogonal Views" function in Imaris and its pipeline for 3D analysis and quantification (e.g., "Spot" module) user friendlier. In this matter, although practically all software tools allow some form of 3D rendering, we recommend Drishti because of its unique light model and shading which enables the generation of highly realistic renderizations (see last segment of Video 4). The rule of thumb for all bioinformatic processing of image datasets (pre-processing and 3D renderings) is that the end result must remain a faithful representation of what is observed in the embryo. Therefore, we provided simple methods to mitigate the problems created by Z-axis scaling distortion (due to refractive index mismatch) and depth signal attenuation (caused by light scattering in deep samples) which severely affect the quality and interpretation of the image datasets. Using these algorithms, we provided instructions to manually segment and 3D reconstruct mesodermal tissues from whole-mount immunofluorescence stainings, by manual contouring (often not possible with tools of automated segmentation) using the Amira software. We find this software especially useful for manual countouring of tissues where there is no clear physical separation or contrast differences between them (e.g., expression of a specific transcription factor). A non-commercial recommended alternative for Amira, although less powerful, is the LabKIT Fiji/ImageJ plugin (https://imagej.net/Labkit).
We have also included among the representative results an example of a 3D visualization of in toto E18.5 mouse fetus imaged with optical projection tomography, which extends the capabilities of optical imaging and 3D image analysis to the entire mouse embryogenesis22. These methodologies can be used to understand and analyze (e.g., through morphological measurements) skeletal abnormalities, like fused vertebrae, scoliosis or spondylocostal dysostosis, that can later occur due to problems during somitogenesis (e.g., Lfng mutation)13, 58,59,60,61,62. Importantly, this imaging approach allows observation of skeletal malformations in a global context, including other tissues (e.g., muscles) that could also contribute to the phenotypes affecting the vertebral column63.
Finally, and in agreement with recent work highlighting volumetric models made with imaging datasets from the eMouse Atlas Project64, we have described a detailed step-by-step method that can be used both to illustrate the power of 3D models (e.g., 3D PDF) to a more general audience and to help in the study and teaching of vertebrate axial elongation and segmentation. We hope the information presented in this manuscript contributes to changing the way researchers design, analyze and present their experiments, and to improve our knowledge of vertebrate body axis formation.
Declarações
The authors have nothing to disclose.
Acknowledgements
We would like to thank Olivier Pourquié and Alexander Aulehla for the LuVeLu reporter strain, the SunJin laboratory for the RapiClear test sample, Hugo Pereira for the help using BigStitcher, Nuno Granjeiro for helping to set up the live imaging apparatus, the IGC animal facility and past and present members of the Mallo lab for useful comments and support during the course of this work.
We thank the technical support of IGC's Advanced Imaging Facility, which is supported by Portuguese funding ref# PPBI-POCI-01-0145-FEDER-022122 and ref# PTDC/BII-BTI/32375/2017, co-financed by Lisboa Regional Operational Programme (Lisboa 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (FEDER) and Fundação para a Ciência e a Tecnologia (FCT, Portugal). Work described in this manuscript was supported by grants LISBOA-01-0145-FEDER-030254 (FCT, Portugal) and SCML-MC-60-2014 (Santa Casa da Misericórdia, Portugal) to M.M., the research infrastructure Congento, project LISBOA-01-0145-FEDER-022170, and the PhD fellowship PD/BD/128426/2017 to A.D.
Materials
| Agarose low gelling temperature | Sigma | A9414 | Used to mounting embryos (e.g. for OPT) |
| Amira software | Thermofisher | – | Commerial software tool |
| Anti-Brachyury (Goat polyclonal) | R and D Systems | AF2085 RRID:AB_2200235 | For immunofluorescence |
| Anti-Sox2 (Rabbit monoclonal) | Abcam | ab92494 RRID:AB_10585428 | For immunofluorescence |
| Anti-Tbx6 (Goat polyclonal) | R and D Systems | AF4744 RRID:AB_2200834 | For immunofluorescence |
| Anti-Laminin111 (Rabbit polyclonal) | Sigma | L9393 RRID:AB_477163 | For immunofluorescence |
| Anti-goat 488 (Donkey polyclonal) | Molecular Probes | A11055 RRID:AB_2534102 | For immunofluorescence |
| Anti-rabbit 568 (Donkey polyclonal) | ThermoFisher Scientific | A10042 RRID:AB_2534017 | For immunofluorescence |
| Benzyl Alcohol (99+%) | (any) | – | Used to clear embryos (component of BABB) |
| Benzyl Benzoate (99+%) | (any) | – | Used to clear embryos (component of BABB) |
| Bovine serum albumin | Biowest | P6154 | For immunofluorescence |
| Coverglass 20×20 mm #0 | (any) | – | 100um thick |
| Coverglass 20×20 mm #1 | (any) | – | 170um thick |
| Coverglass 20×60 mm #1.5 | (any) | – | To use as “slides” |
| DAPI (4’,6-Diamidino-2- Phenylindole Dihydrochloride) | Life Technologies | D3571 | For immunofluorescence |
| Drishti software | (open source) | – | Free software tool |
| EDTA | Sigma | ED2SS | For demineralization |
| Fiji/ImageJ software | (open source) | – | Free software tool |
| Glycine | NZYtech | MB01401 | For immunofluorescence |
| Huygens software | Scientific Volume Imaging | – | Commerial software tool |
| HyClone defined fetal bovine serum | GE Healthcare | #HYCLSH30070.03 | For live imaging |
| Hydrogen peroxide solution 30 % | Milipore | 1085971000 | For clearing |
| Imaris software | Bitplane / Oxford instruments | – | Commerial software tool |
| iSpacers | SunJin Lab | (varies) | Use as spacers for preparations |
| L-glutamine | Gibco | #25030–024 | For live imaging medium |
| Low glucose DMEM | Gibco | 11054020 | For live imaging medium |
| M2 medium | Sigma | M7167 | To dissect embryos |
| Methanol | VWR | VWRC20847.307 | For dehydration and rehydration steps |
| Methyl salicylate | Sigma | M6752 | Used to clear embryos |
| Paraformaldehyde | Sigma | P6148 | Used in solution to fix embryos |
| Penicillin-streptomycin | Sigma | #P0781 | For live imaging medium |
| PBS (Phosphate-buffered saline solution) | Biowest | L0615-500 | – |
| RapiClear | SunJin Laboratory | RapiClear 1.52 | Used to clear embryos |
| Secure-Sea hybridization chambers | Sigma | C5474 | Use as spacers for preparations |
| simLab software | SimLab soft | – | Commerial software tool |
| Slide, depression concave glass – 75×25 mm | (any) | – | To mount thick embryos. |
| Triton X-100 | Sigma | T8787 | For immunofluorescence |
Referências
- Wilson, V., Olivera-Martinez, I., Storey, K. G. Stem cells, signals and vertebrate body axis extension. Development. 136 (12), 2133 (2009).
- Dias, A., Aires, R. Axial Stem Cells and the Formation of the Vertebrate Body. Concepts and Applications of Stem Cell Biology. , 131-158 (2020).
- Tzouanacou, E., Wegener, A., Wymeersch, F. J., Wilson, V., Nicolas, J. F. Redefining the Progression of Lineage Segregations during Mammalian Embryogenesis by Clonal Analysis. Developmental Cell. 17 (3), 365-376 (2009).
- Aires, R., Dias, A., Mallo, M. Deconstructing the molecular mechanisms shaping the vertebrate body plan. Current Opinion in Cell Biology. 55, 81-86 (2018).
- Wymeersch, F. J., et al. Position-dependent plasticity of distinct progenitor types in the primitive streak. eLife. 5, 10042 (2016).
- Koch, F., et al. Antagonistic Activities of Sox2 and Brachyury Control the Fate Choice of Neuro-Mesodermal Progenitors. Developmental Cell. 42 (5), 514-526 (2017).
- Chapman, D. L., Papaioannou, V. E. Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature. 391 (1991), 695-697 (1998).
- de Lemos, L., Dias, A., Nóvoa, A., Mallo, M. Epha1 is a cell surface marker for neuromesodermal progenitors and their early mesoderm derivatives. bioRxiv. , 584524 (2020).
- Takada, S., Stark, K. L., Shea, M. J., Vassileva, G., McMahon, J. A., McMahon, A. P. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes and Development. 8 (2), 174-189 (1994).
- Chalamalasetty, R. B., et al. Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development. 141 (22), 4285-4297 (2014).
- Pais-de-Azevedo, T., Magno, R., Duarte, I., Palmeirim, I. Recent advances in understanding the mechanisms determining longevity. F1000Research. 7, 97 (2018).
- Hubaud, A., Pourquié, O. Signalling dynamics in vertebrate segmentation. Nature Reviews Molecular Cell Biology. 15 (11), 709-721 (2014).
- Pourquié, O. Vertebrate segmentation: From cyclic gene networks to scoliosis. Cell. 145 (5), 650-663 (2011).
- Mallo, M. Revisiting the involvement of signaling gradients in somitogenesis. FEBS Journal. 283 (8), 1430-1437 (2016).
- Boulet, A. M., Capecchi, M. R. Signaling by FGF4 and FGF8 is required for axial elongation of the mouse embryo. Biologia do Desenvolvimento. 371 (2), 235-245 (2012).
- Dickinson, M. E., et al. High-throughput discovery of novel developmental phenotypes. Nature. 537 (7621), 508-514 (2016).
- McDole, K., et al. In toto Imaging and Reconstruction of Post-Implantation Mouse Development at the Single-Cell Level. Cell. 175, 859-876 (2018).
- McColl, J., Mok, G. F., Lippert, A. H., Ponjavic, A., Muresan, L., Münsterberg, A. 4D imaging reveals stage dependent random and directed cell motion during somite morphogenesis. Scientific Reports. 8 (1), 12644 (2018).
- Martins, G. G., Rifes, P., Amaândio, R., Rodrigues, G., Palmeirim, I., Thorsteinsdóttir, S. Dynamic 3D cell rearrangements guided by a fibronectin matrix underlie somitogenesis. PLoS ONE. 4 (10), 7429 (2009).
- Bénazéraf, B., et al. Multi-scale quantification of tissue behavior during amniote embryo axis elongation. Development. 144, 4462-4472 (2017).
- Sharpe, J. Optical Projection Tomography. Advanced Imaging in Biology and Medicine. , 199-224 (2009).
- Sharpe, J., et al. Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science. 296 (5567), 541-545 (2002).
- Aulehla, A., et al. A beta-catenin gradient links the clock and wavefront systems in mouse embryo segmentation. Nature Cell Biology. 10 (2), 186-193 (2008).
- Osorno, R., et al. The developmental dismantling of pluripotency is reversed by ectopic Oct4 expression. Development. 139 (13), 2288-2298 (2012).
- Bryson-Richardson, R. J., Currie, P. D. Optical projection tomography for spatio-temporal analysis in zebrafish. Methods in Cell Biology. 76, 37-50 (2004).
- Quintana, L., Sharpe, J. Optical projection tomography of vertebrate embryo development. Cold Spring Harbor Protocols. 6 (6), 586-594 (2011).
- Cho, A., Suzuki, S., Hatakeyama, J., Haruyama, N., Kulkarni, A. B. A Method for Rapid Demineralization of Teeth and Bones. The Open Dentistry Journal. 4 (1), 223-229 (2010).
- Gualda, E. J., Vale, T., Almada, P., Feijó, J. A., Martins, G. G., Moreno, N. OpenSpinMicroscopy: An open-source integrated microscopy platform. Nature Methods. 10 (7), 599-600 (2013).
- Schindelin, J., et al. Fiji: An open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Weigert, M., et al. Content-aware image restoration: pushing the limits of fluorescence microscopy. Nature Methods. 15 (12), 1090-1097 (2018).
- Krull, A., Buchholz, T. O., Jug, F. Noise2void-learning denoising from single noisy images. 2019 IEEE. CVF Conference on Computer Vision and Pattern Recognition (CVPR). , 2124-2132 (2018).
- Sage, D., et al. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods. 115, 28-41 (2017).
- Pietzsch, T., Saalfeld, S., Preibisch, S., Tomancak, P. BigDataViewer: Visualization and processing for large image data sets. Nature Methods. 12 (6), 481-483 (2015).
- Parslow, A., Cardona, A., Bryson-Richardson, R. J. Sample Drift Correction Following 4D Confocal Time-lapse Imaging. Journal of Visualized Experiments. (86), e51086 (2014).
- Hörl, D., et al. BigStitcher: reconstructing high-resolution image datasets of cleared and expanded samples. Nature Methods. 16, 870-874 (2019).
- Ohser, J., et al. Attenuation correction for confocal laser scanning microscopy and its application in chromatography. Journal of Microscopy. 278, 76-88 (2020).
- Hell, S., Reiner, G., Cremer, C., Stelzer, E. H. K. Aberrations in confocal fluorescence microscopy induced by mismatches in refractive index. Journal of Microscopy. 169, 391-405 (1993).
- Kuypers, L. C., Decraemer, W. F., Dirckx, J. J. J., Timmermans, J. A procedure to determine the correct thickness of an object with confocal microscopy in case of refractive index mismatch. Journal of Microscopy. 218, 68-78 (2005).
- Meijering, E. H. W., Niessen, W. J., Viergever, M. A. Quantitative evaluation of convolution-based methods for medical image interpolation. Medical Image Analysis. 5 (2), 111-126 (2001).
- Limaye, A. Drishti: a volume exploration and presentation tool. Developments in X-Ray Tomography VIII. , 85060 (2012).
- . Thermofisher.com Available from: https://www.thermofisher.com/pt/en/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software/3d-visualization-analysis-software-resource-center.html (2020)
- . Simlab-soft.com Available from: https://www.simlab-soft.com/3d-products/Tutorials/simlab-composer-all-Tutorials.aspx?t=3 (2020)
- Dias, A., et al. A TgfbRI/Snai1-dependent developmental module at the core of vertebrate axial elongation. eLife. 9, 56615 (2020).
- Delaune, E. A., François, P., Shih, N. P., Amacher, S. L. Single-Cell-Resolution Imaging of the Impact of Notch Signaling and Mitosis on Segmentation Clock Dynamics. Developmental Cell. 23 (5), 995-1005 (2012).
- Attardi, A., et al. Neuromesodermal progenitors are a conserved source of spinal cord with divergent growth dynamics. Development. 145 (21), (2018).
- Romanos, M., et al. Cell-to-cell heterogeneity in Sox2 and Brachyury expression guides progenitor destiny by controlling their movements. bioRxiv. , 388611 (2020).
- Guillot, C., Michaut, A., Rabe, B., Pourquié, O. Dynamics of primitive streak regression controls the fate of neuro-mesodermal progenitors in the chicken embryo. bioRxiv. , 077586 (2020).
- Steventon, B., Duarte, F., Lagadec, R., Mazan, S., Nicolas, J. F., Hirsinger, E. Species-specific contribution of volumetric growth and tissue convergence to posterior body elongation in vertebrates. Development. 143 (10), 1732-1741 (2016).
- Lawton, A. K., et al. Regulated tissue fluidity steers zebrafish body elongation. Development. 140 (3), 573-582 (2013).
- Huang, Q., et al. Intravital imaging of mouse embryos. Science. 368 (6487), 181-186 (2020).
- Van Den Brink, S. C., et al. Symmetry breaking, germ layer specification and axial organisation in aggregates of mouse embryonic stem cells. Development. 141 (22), 4231-4242 (2014).
- Baillie-Johnson, P., vanden Brink, S. C., Balayo, T., Turner, D. A., Martinez Arias, A. Generation of aggregates of mouse embryonic stem cells that show symmetry breaking, polarization and emergent collective behaviour in vitro. Journal of Visualized Experiments. (105), e53252 (2015).
- Moris, N., et al. An in vitro model of early anteroposterior organization during human development. Nature. 582, 410-415 (2020).
- Rossner, M., Yamada, K. M. What’s in a picture? The temptation of image manipulation. The Journal of Cell Biology. 166 (1), 11-15 (2004).
- North, A. J. Seeing is believing? A beginners’ guide to practical pitfalls in image acquisition. Journal of Cell Biology. 172 (1), 9-18 (2006).
- Jost, A. P. -. T., Waters, J. C. Designing a rigorous microscopy experiment: Validating methods and avoiding bias. Journal of Cell Biology. 218 (5), 1452-1466 (2019).
- Jonkman, J., Brown, C. M., Wright, G. D., Anderson, K. I., North, A. J. Tutorial: guidance for quantitative confocal microscopy. Nature Protocols. 15 (5), 1585-1611 (2020).
- Stauber, M., Sachidanandan, C., Morgenstern, C., Ish-Horowicz, D. Differential axial requirements for Lunatic fringe and Hes7 transcription during mouse somitogenesis. PLoS ONE. 4 (11), 7996 (2009).
- Turnpenny, P. D., et al. Abnormal vertebral segmentation and the notch signaling pathway in man. Developmental Dynamics. 236 (6), 1456-1474 (2007).
- Andrade, R. P., Palmeirim, I., Bajanca, F. Molecular clocks underlying vertebrate embryo segmentation: A 10-year-old hairy-go-round. Birth Defects Research (Part C). 81 (2), 65-83 (2007).
- Sparrow, D. B., et al. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. American Journal of Human Genetics. 78 (1), 28-37 (2006).
- Giampietro, P. F., et al. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Annals of the New York Academy of Sciences. 1151, 38-67 (2009).
- Blecher, R., et al. The Proprioceptive System Masterminds Spinal Alignment: Insight into the Mechanism of Scoliosis. Developmental Cell. 42 (4), 388-399 (2017).
- Vianello, S. Exploring and illustrating the mouse embryo virtual objects to think and create with. bioRxiv. , 393991 (2020).
