In den letzten zwei Jahrzehnten haben sich Biotherapeutika zu einer tragenden Säule der modernen Pharmaindustrie entwickelt. Die SARS-CoV2-Pandemie und andere lebensbedrohliche Erkrankungen haben den Bedarf an einer schnelleren und breiteren Entwicklung biopharmazeutischer Moleküle weiter erhöht 1,2,3.
Das biotherapeutische Molekulargewicht ist in Kombination mit anderen analytischen Assays entscheidend für die Identifizierung des Moleküls. Die intakten und reduzierten Untereinheitenmassen werden während des gesamten Forschungs- und Entwicklungslebenszyklus als Teil von Kontrollstrategien verwendet, die darauf abzielen, die Qualität aufrechtzuerhalten, wie im QTPP (Quality Target Product Profile)4 beschrieben.
Die analytische Entwicklung in der biopharmazeutischen Industrie stützt sich in hohem Maße auf Massenmessungen für die Analyse intakter Massen und eine tiefgreifende Charakterisierung mittels Peptid-Mapping oder Multi-Attribut-Methode (MAM)-Überwachung. Im Mittelpunkt dieser Techniken, bei denen moderne Massenspektrometrie-Plattformen (MS) zum Einsatz kommen, steht die Fähigkeit, hochauflösende, genaue Massenmessungen (HR/AM) durchzuführen. Die meisten HR/AM-Geräte liefern Massengenauigkeiten im Bereich von 0,5 bis 5 ppm, die mit dem Massenbereich skaliert werden. Die Fähigkeit, Massen für intakte große Moleküle genau zu messen, ermöglicht die schnelle und sichere Identifizierung von Therapeutika mit großen Molekülen. Da die Isotopenauflösung unter typischen experimentellen Bedingungen für große Moleküle (>10 kDa) nicht erreicht werden kann, müssen für den Vergleich und die Identifizierung mittlere Massen berechnet werden 5,6.
Ein typisches Massenspektrum eines intakten oder untergliedrigen Proteins stellt das gesamte Proteoformprofil dar, das zusammengesetzte Informationen über die verschiedenen molekularen Formen enthält, die sich aus posttranslationalen Modifikationen (PTM) und etwaigen primären Strukturunterschieden, wie z. B. Clips oder Sequenzvarianten, ergeben. Die relativ einfache Natur und der hohe Durchsatz dieser Messungen machen sie attraktiv für die Charakterisierung und als In-Prozess-Überwachungskontrollen 7,8. Die Datenanalyse für diese Experimente erfordert in der Regel, dass der Benutzer den Suchraum für molekulare Formen (Bereich von PTMs oder anderen molekularen Formen) definiert. Für glykosylierte Proteine wird dieser Suchraum weitgehend durch die Heterogenität der Glykoformen bestimmt. Kombinationen aus mehreren PTMs, Disulfidbrückenkonfigurationen und anderen Variationen entlang der Primärstruktur machen die Berechnung aller möglichen Molekülformen zu einer mühsamen Aufgabe. Daher ist die manuelle Berechnung der möglichen Molekülformen ein zeit- und ressourcenintensiver Prozess mit einem hohen Potenzial für menschliche Fehler.
Hier stellen wir ein Massenberechnungstool vor, das unter Berücksichtigung der wichtigsten Eigenschaften biotherapeutischer Moleküle, wie z.B. mAbs, bsAbs, ADCs, etc., entwickelt wurde. Das Werkzeug ermöglicht die einfache Einbindung von Suchraumvariablen zur konsistenten Berechnung von Massen und Elementzusammensetzungen. Der modulare Charakter dieses Werkzeugs wird es ermöglichen, es weiterzuentwickeln und auf die Massenberechnung und das Massenanpassungssystem für andere Modalitäten anzuwenden.
Das GUI-Modul ermöglicht es dem Benutzer, die Eingabe für die Massenberechnung anzugeben, wie in Abbildung 1 gezeigt. Konkret gibt der Anwender einbuchstabige Aminosäuresequenzen für leichte und schwere Antikörperketten ein. Gängige Modifikationen für die N-terminale Zyklisierung mit schweren Ketten und das C-terminale Lysin-Clipping sind als Kontrollkästchen enthalten. Ferner kann die chemische Formel/Elementzusammensetzung über das entsprechende Chem Mod-Textfeld zu diesen Proteinketten addiert/subtrahiert werden. Dies gibt dem Benutzer die Flexibilität, eine elementare Zusammensetzung hinzuzufügen, die mehrere posttranslationale Modifikationen oder im Falle eines ADC eine kleinmolekulare Nutzlast enthält. Da die meisten therapeutischen mAbs so konstruiert sind, dass sie die Glykosylierungsstellen in der Leichtkette entfernen, ist die Glykosylierung in der Leichtkette optional und kann über ein Kontrollkästchen in der GUI angegeben werden.
Eine typische Variante der Analyse intakter Massen für Antikörper ist eine Analyse mit reduzierter Untereinheitsmasse, bei der die leichte Kette von der schweren Kette gelöst wird, indem die Disulfidbrücken zwischen den Ketten reduziert werden. Abhängig von der Stärke des verwendeten Reduktionsmittels können die Disulfidbindungen innerhalb der Kette gespalten werden oder nicht. Die Benutzer haben die Flexibilität, die Gesamtzahl der Disulfidbrücken in Abhängigkeit vom IgG-Subtyp oder im Falle eines Cystein-konjugierten ADC9 einzugeben.
Die Anwendung berechnet Massen nach dem Bottom-up-Verfahren, wobei zunächst die elementaren Zusammensetzungen für die einzelnen schweren Ketten und leichten Ketten berechnet werden. Als nächstes wird das Lys-Clipping der Schwerketten-N-terminalen Cyclisierung (HC) durch Anpassung der berechneten Elementzusammensetzungen berücksichtigt. Alle spezifizierten chemischen Modifikationen werden dann auf die schweren und/oder leichten Ketten aufgebracht. Abhängig von der Art der Analyse und den vom Anwender vorgegebenen Disulfidbindungsmustern wird die Anzahl der Wasserstoffe für die beiden Polypeptidketten angepasst. Die glykosylierten HC- und Leichtkettenmassen (LC) (optional) werden auf der Grundlage der Eingaben des Benutzers berechnet. Schließlich werden mehrere HC- und LC-Massen kombiniert, und die Disulfidbrückennummern werden automatisch für die Berechnung der intakten Masse aktualisiert.
Bei größeren Molekülen, wie z.B. intakten Proteinen, können monoisotopische Massen aufgrund des additiven Massendefekts bei der Verwendung von Massenspektrometern mit typischem Auflösungsvermögen nicht gemessen werden. Stattdessen werden Nenn- oder Durchschnittsmassen gemessen oder gemeldet 5,10,11,12,13. Die durchschnittlichen Elementmassen können je nach der Quelle, die für die kuratierten Massen verwendet wird, variieren14,15. Die Unterschiede in den Elementmassen mögen zwar gering sein, können sich aber für die Berechnung des Molekulargewichts großer Moleküle zu signifikanten Werten summieren. Die durchschnittlichen Elementmassen, die standardmäßig in der Softwareanwendung verwendet werden, sind in der ergänzenden Tabelle 1 aufgeführt. Für regulierte Umgebungen wie den Bereich der biopharmazeutischen Forschung und Entwicklung (F&E) ist es wichtig, konsistente Molekülmassen beizubehalten, da Änderungen der Massen Änderungen an der molekularen Einheit während der Zulassungsanträge bedeuten können. Um eine konsistente Verwendung von Elementmassen zu ermöglichen, ist ein Wörterbuch der Elementmassen als CSV-Textdatei (Comma-Separated Value) im Software-Tool enthalten: Element_Mass.csv (Supplementary Coding File 1). In ähnlicher Weise ist eine kuratierte Liste von Glykanzusammensetzungen enthalten, die typischerweise auf mAbs zu sehen sind: Glycan.csv (Supplementary Coding File 2). Beide Dateien werden im selben Ordner wie eine ausführbare Anwendung gespeichert und können vom Benutzer geändert werden, um eine bestimmte Elementmassenliste oder Glykanbibliothek zu verwenden.
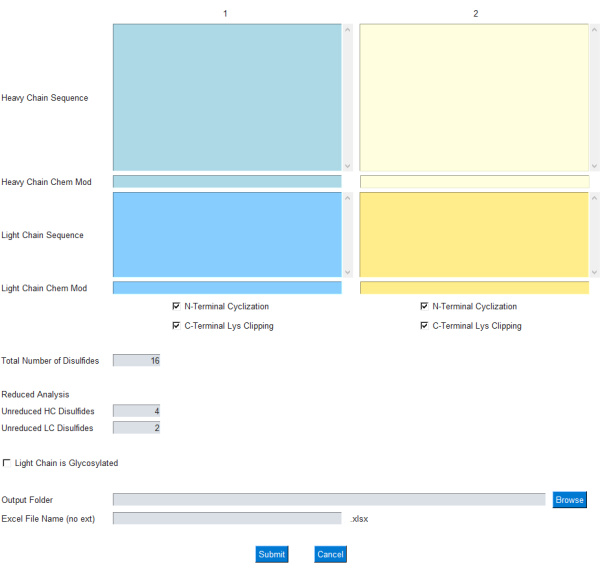
Abbildung 1: GUI-Schnittstelle für die mAbScale-Anwendung. Das GUI-Modul ermöglicht es dem Benutzer, die Eingabe für die Massenberechnung anzugeben. Der Benutzer gibt einbuchstabige Aminosäuresequenzen für die leichten und schweren Antikörperketten ein. Gängige Modifikationen für die N-terminale Zyklisierung mit schweren Ketten und das C-terminale Lysin-Clipping sind als Kontrollkästchen enthalten. Chemische Formeln/Elementzusammensetzungen können über das entsprechende Chem Mod-Textfeld addiert/subtrahiert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
