In de afgelopen twee decennia zijn biotherapeutica geëvolueerd tot een steunpilaar van de moderne farmaceutische industrie. De SARS-CoV2-pandemie en andere levensbedreigende omstandigheden hebben de behoefte aan een snellere en bredere ontwikkeling van biofarmaceutische moleculen verder vergroot 1,2,3.
Het biotherapeutische molecuulgewicht is van cruciaal belang voor de identificatie van het molecuul, in combinatie met andere analytische tests. De intacte en gereduceerde subeenheidmassa’s worden gedurende de hele ontdekkings- en ontwikkelingscyclus gebruikt als onderdeel van controlestrategieën die gericht zijn op het handhaven van de kwaliteit, zoals beschreven in het QTPP (Quality Target Product Profile)4.
Analytische ontwikkeling in de biofarmaceutische industrie is sterk afhankelijk van massametingen voor intacte massa-analyse en diepe karakterisering met behulp van peptidemapping of multi-attribute method (MAM) monitoring. Centraal in deze technieken die gebruik maken van moderne massaspectrometrieplatforms (MS) staat de mogelijkheid om nauwkeurige massametingen (HR/AM) met hoge resolutie te leveren. De meeste HR/AM-instrumenten leveren massanauwkeurigheden in het bereik van 0,5-5 ppm, die schalen met het massabereik. Het vermogen om massa’s nauwkeurig te meten voor intacte grote moleculen maakt de snelle en betrouwbare identificatie van therapieën met grote moleculen mogelijk. Aangezien de isotopenresolutie niet kan worden bereikt onder typische experimentele omstandigheden voor grote moleculen (>10 kDa), moeten de gemiddelde massa’s worden berekend voor vergelijking en identificatie 5,6.
Een typisch intact of subeenheid eiwitmassaspectrum vertegenwoordigt het algemene proteoforme profiel, dat samengestelde informatie bevat over de verschillende moleculaire vormen die het gevolg zijn van posttranslationele modificaties (PTM) en eventuele verschillen in de primaire structuur, zoals clips of sequentievarianten. Het relatief eenvoudige en hoge doorvoerkarakter van deze metingen maakt ze aantrekkelijk voor karakterisering en als controlemechanismen tijdens het proces 7,8. Data-analyse voor deze experimenten vereist meestal dat de gebruiker de zoekruimte voor moleculaire vormen (bereik van PTM’s of andere moleculaire vormen) definieert. Voor geglycosyleerde eiwitten wordt deze zoekruimte grotendeels bepaald door glycoforme heterogeniteit. Combinaties van meerdere PTM’s, disulfidebindingsconfiguraties en andere variaties langs de primaire structuur maken het berekenen van alle mogelijke moleculaire vormen een vervelende taak. Daarom is de handmatige berekening van de mogelijke moleculaire vormen een tijdrovend en tijdrovend proces met een grote kans op menselijke fouten.
Hier presenteren we een massaberekeningstool die is ontwikkeld rekening houdend met de belangrijkste kenmerken van biotherapeutische moleculen, zoals mAbs, bsAbs, ADC’s, enz. De tool maakt het mogelijk om eenvoudig zoekruimtevariabelen op te nemen voor de consistente berekening van massa’s en elementaire samenstellingen. Het modulaire karakter van deze tool zal het mogelijk maken om deze verder te ontwikkelen en toe te passen op massaberekening en massa-matching voor andere modaliteiten.
De GUI-module stelt de gebruiker in staat om de invoer voor de massaberekening te specificeren, zoals weergegeven in figuur 1; In het bijzonder voert de gebruiker aminozuursequenties van één letter in voor lichte en zware antilichaamketens. Veelvoorkomende modificaties voor zware keten N-terminale cyclisatie en C-terminale lysineclipping zijn opgenomen als selectievakjes. Verder kan de chemische formule/elementaire samenstelling worden opgeteld/afgetrokken van deze eiwitketens via het respectievelijke Chem Mod-tekstvak . Dit geeft de gebruiker de flexibiliteit om een elementaire samenstelling toe te voegen die meerdere posttranslationele modificaties of een kleine molecuullading bevat in het geval van een ADC. Aangezien de meeste therapeutische mAbs zijn ontworpen om de glycosyleringsplaatsen in de lichte keten te verwijderen, wordt glycosylering in de lichte keten optioneel gelaten en kan deze worden gespecificeerd met behulp van een selectievakje op de GUI.
Een typische variatie op intacte massa-analyse voor antilichamen is een verminderde subeenheidmassa-analyse, waarbij de lichte keten wordt losgemaakt van de zware keten door de disulfidebindingen tussen de ketens te verminderen. Afhankelijk van de sterkte van het gebruikte reductiemiddel kunnen de disulfidebindingen binnen de keten al dan niet worden gesplitst. De gebruikers hebben de flexibiliteit om het totale aantal disulfidebindingen in te voeren, afhankelijk van het IgG-subtype of in het geval van een cysteïne-geconjugeerde ADC9.
De applicatie berekent massa’s op een bottom-up manier, waarbij eerst de elementaire samenstellingen worden berekend voor de individuele zware ketens en lichte ketens. Vervolgens wordt rekening gehouden met zware keten (HC) N-terminale cyclisatie Lys-clipping door de berekende elementaire samenstellingen aan te passen. Eventuele gespecificeerde chemische modificaties worden vervolgens toegepast op de zware en/of lichte kettingen. Afhankelijk van het type analyse en de door de gebruiker gespecificeerde disulfidebindingspatronen, wordt het aantal waterstofatomen aangepast voor de twee polypeptideketens. De geglycosyleerde HC en lichte keten (LC) (optioneel) massa’s worden berekend op basis van de input van de gebruiker. Ten slotte worden meerdere HC- en LC-massa’s gecombineerd en worden de disulfidebindingsnummers automatisch bijgewerkt voor de berekening van de intacte massa.
Bij grotere moleculen, zoals intacte eiwitten, kunnen mono-isotopische massa’s niet worden gemeten vanwege het additieve massadefect bij gebruik van massaspectrometers met een typisch oplossend vermogen. In plaats daarvan worden nominale of gemiddelde massa’s gemeten of gerapporteerd 5,10,11,12,13. De gemiddelde elementaire massa’s kunnen variëren op basis van de bron die wordt gebruikt voor de samengestelde massa’s14,15. Hoewel de verschillen in elementaire massa’s klein kunnen zijn, kunnen ze oplopen tot significante waarden voor berekeningen van het molecuulgewicht van grote moleculen. De gemiddelde elementaire massa’s die standaard in de softwaretoepassing worden gebruikt, worden weergegeven in aanvullende tabel 1. Voor gereguleerde omgevingen zoals het biofarmaceutische onderzoeks- en ontwikkelingsveld (R&D) is het belangrijk om consistente molecuulmassa’s te behouden, omdat veranderingen in massa’s veranderingen in de moleculaire entiteit kunnen impliceren tijdens regelgevende deponeringen. Om consistentie in het gebruik van elementaire massa’s mogelijk te maken, is een woordenboek van elementaire massa’s bij de softwaretool opgenomen als een door komma’s gescheiden waarde (csv) tekstbestand: Element_Mass.csv (Supplementary Coding File 1). Evenzo is een samengestelde lijst van glycaansamenstellingen opgenomen die typisch zijn voor mAbs: Glycaan.csv (Supplementary Coding File 2). Beide bestanden worden opgeslagen op dezelfde maplocatie als een uitvoerbare toepassing en kunnen door de gebruiker worden gewijzigd om een specifieke elementaire massalijst of glycaanbibliotheek te gebruiken.
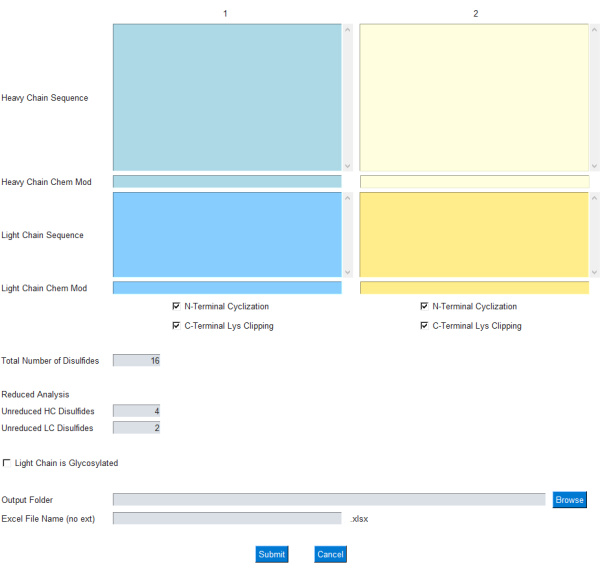
Figuur 1: GUI-interface voor de mAbScale-applicatie. De GUI-module stelt de gebruiker in staat om de invoer voor de massaberekening te specificeren. De gebruiker voert aminozuursequenties van één letter in voor de lichte en zware antilichaamketens. Veelvoorkomende modificaties voor de zware-keten N-terminale cyclisatie en C-terminale lysineclipping zijn opgenomen als selectievakjes. Chemische formules/elementaire samenstellingen kunnen worden opgeteld/afgetrokken via het respectievelijke Chem Mod-tekstvak . Klik hier om een grotere versie van deze figuur te bekijken.
