पिछले दो दशकों में, बायोथेरेप्यूटिक्स आधुनिक दवा उद्योग का मुख्य आधार बनने के लिए विकसित हुए हैं। सार्स-सीओवी2 महामारी और अन्य जानलेवा स्थितियों ने बायोफार्मास्युटिकलअणुओं 1,2,3 के तेज और व्यापक विकास की आवश्यकता को और बढ़ा दिया है।
अन्य विश्लेषणात्मक परखों के साथ संयोजन में, अणु की पहचान के लिए बायोथेराप्यूटिक आणविक भार महत्वपूर्ण है। गुणवत्ता बनाए रखने के उद्देश्य से नियंत्रण रणनीतियों के हिस्से के रूप में खोज और विकास जीवनचक्र में बरकरार और कम सबयूनिट द्रव्यमान का उपयोग किया जाता है, जैसा कि क्यूटीपीपी (गुणवत्ता लक्ष्य उत्पाद प्रोफ़ाइल) 4 में वर्णित है।
बायोफार्मास्युटिकल उद्योग में विश्लेषणात्मक विकास पेप्टाइड मैपिंग या मल्टी-एट्रिब्यूट विधि (एमएएम) निगरानी का उपयोग करके बरकरार द्रव्यमान विश्लेषण और गहरे लक्षण वर्णन के लिए बड़े पैमाने पर माप पर बहुत अधिक निर्भर करता है। आधुनिक मास स्पेक्ट्रोमेट्री (एमएस) प्लेटफार्मों का उपयोग करने वाली इन तकनीकों के केंद्र में उच्च-रिज़ॉल्यूशन सटीक द्रव्यमान (एचआर / एएम) माप प्रदान करने की क्षमता है। अधिकांश एचआर / एएम उपकरण 0.5-5 पीपीएम की सीमा में द्रव्यमान सटीकता उत्पन्न करते हैं, जो द्रव्यमान सीमा के साथ स्केल करते हैं। बरकरार बड़े अणुओं के लिए द्रव्यमान को सटीक रूप से मापने की क्षमता बड़े अणु चिकित्सीय की त्वरित और आत्मविश्वास ी पहचान को सक्षम बनाती है। चूंकि आइसोटोपिक रिज़ॉल्यूशन बड़े अणुओं (>10 केडीए) के लिए विशिष्ट प्रयोगात्मक स्थितियों का उपयोग करके प्राप्त नहीं किया जा सकता है, औसत द्रव्यमान की गणना तुलना और पहचान 5,6 के लिए की जानी चाहिए।
एक विशिष्ट बरकरार या सबयूनिट प्रोटीन मास स्पेक्ट्रम समग्र प्रोटियोफोम प्रोफाइल का प्रतिनिधित्व करता है, जिसमें पोस्ट-ट्रांसलेशनल संशोधनों (पीटीएम) और किसी भी प्राथमिक संरचना अंतर, जैसे क्लिप या अनुक्रम वेरिएंट के परिणामस्वरूप विभिन्न आणविक रूपों पर समग्र जानकारी होती है। इन मापों की अपेक्षाकृत आसान और उच्च-थ्रूपुट प्रकृति उन्हें लक्षण वर्णन के लिए आकर्षक बनाती है और इन-प्रोसेस मॉनिटरिंग कंट्रोल 7,8 के रूप में। इन प्रयोगों के लिए डेटा विश्लेषण के लिए आमतौर पर उपयोगकर्ता को आणविक रूपों (पीटीएम या अन्य आणविक रूपों की सीमा) के लिए खोज स्थान को परिभाषित करने की आवश्यकता होती है। ग्लाइकोसिलेटेड प्रोटीन के लिए, यह खोज स्थान काफी हद तक ग्लाइकोफॉर्म विषमता से प्रेरित है। प्राथमिक संरचना के साथ कई पीटीएम, डाइसल्फ़ाइड बॉन्ड कॉन्फ़िगरेशन और अन्य विविधताओं के संयोजन सभी संभावित आणविक रूपों की गणना करना एक थकाऊ काम बनाते हैं। इसलिए, संभावित आणविक रूपों की मैन्युअल गणना मानव त्रुटि के लिए उच्च क्षमता के साथ एक समय और संसाधन लेने वाली प्रक्रिया है।
यहां, हम एक बड़े पैमाने पर गणना उपकरण प्रस्तुत करते हैं जिसे बायोथेराप्यूटिक अणुओं की सबसे महत्वपूर्ण विशेषताओं पर विचार करते हुए विकसित किया गया था, जैसे कि एमएबीएस, बीएसएबीएस, एडीसी, आदि। उपकरण द्रव्यमान और मौलिक रचनाओं की सुसंगत गणना के लिए खोज-स्थान चर के आसान समावेश की अनुमति देता है। इस उपकरण की मॉड्यूलर प्रकृति इसे आगे विकसित करने और अन्य तौर-तरीकों के लिए बड़े पैमाने पर गणना और द्रव्यमान मिलान पर लागू करने में सक्षम करेगी।
जीयूआई मॉड्यूल उपयोगकर्ता को द्रव्यमान गणना के लिए इनपुट निर्दिष्ट करने की अनुमति देता है, जैसा कि चित्रा 1 में दिखाया गया है; विशेष रूप से, उपयोगकर्ता हल्के और भारी एंटीबॉडी श्रृंखलाओं के लिए एकल-अक्षर अमीनो एसिड अनुक्रम दर्ज करता है। भारी-श्रृंखला एन-टर्मिनल साइक्लाइजेशन और सी-टर्मिनल लाइसिन क्लिपिंग के लिए सामान्य संशोधन चेक बॉक्स के रूप में शामिल हैं। इसके अलावा, रासायनिक सूत्र/मौलिक संरचना को संबंधित केम मॉड टेक्स्ट बॉक्स के माध्यम से इन प्रोटीन श्रृंखलाओं से जोड़ा/घटाया जा सकता है। यह उपयोगकर्ता को एक मौलिक संरचना जोड़ने की सुविधा देता है जिसमें एडीसी के मामले में कई पोस्ट-ट्रांसलेशनल संशोधन या एक छोटा-अणु पेलोड शामिल है। चूंकि अधिकांश चिकित्सीय एमएबी को प्रकाश श्रृंखला में ग्लाइकोसिलेशन साइटों को हटाने के लिए इंजीनियर किया जाता है, इसलिए प्रकाश श्रृंखला में ग्लाइकोसिलेशन को वैकल्पिक छोड़ दिया जाता है और जीयूआई पर चेक बॉक्स का उपयोग करके निर्दिष्ट किया जा सकता है।
एंटीबॉडी के लिए बरकरार द्रव्यमान विश्लेषण पर एक विशिष्ट भिन्नता एक कम सबयूनिट द्रव्यमान विश्लेषण है, जहां इंटरचेन डाइसल्फ़ाइड बॉन्ड को कम करके प्रकाश श्रृंखला को भारी श्रृंखला से अलग किया जाता है। उपयोग किए गए कम करने वाले एजेंट की ताकत के आधार पर, इंट्राचेन डाइसल्फ़ाइड बॉन्ड को क्लीवर किया जा सकता है या नहीं। उपयोगकर्ताओं के पास आईजीजी उपप्रकार के आधार पर या सिस्टीन-संयुग्मित एडीसी9 के मामले में डाइसल्फ़ाइड बॉन्ड की कुल संख्या दर्ज करने की लचीलापन है।
एप्लिकेशन एक नीचे-ऊपर तरीके से द्रव्यमान की गणना करता है, जिसमें मौलिक रचनाओं की गणना पहले व्यक्तिगत भारी श्रृंखलाओं और प्रकाश श्रृंखलाओं के लिए की जाती है। इसके बाद, भारी श्रृंखला (एचसी) एन-टर्मिनल साइक्लाइजेशन लिस-क्लिपिंग को गणना की गई मौलिक रचनाओं को समायोजित करके हिसाब लगाया जाता है। किसी भी निर्दिष्ट रासायनिक संशोधनों को तब भारी और / या हल्की श्रृंखलाओं पर लागू किया जाता है। विश्लेषण के प्रकार और उपयोगकर्ता द्वारा निर्दिष्ट डाइसल्फ़ाइड-बॉन्ड पैटर्न के आधार पर, हाइड्रोजन की संख्या को दो पॉलीपेप्टाइड श्रृंखलाओं के लिए समायोजित किया जाता है। ग्लाइकोसिलेटेड एचसी और लाइट चेन (एलसी) (वैकल्पिक) द्रव्यमान की गणना उपयोगकर्ता के इनपुट के आधार पर की जाती है। अंत में, कई एचसी और एलसी द्रव्यमान संयुक्त होते हैं, और बरकरार द्रव्यमान गणना के लिए डाइसल्फ़ाइड बॉन्ड नंबर स्वचालित रूप से अपडेट किए जाते हैं।
बरकरार प्रोटीन जैसे बड़े अणुओं के साथ, विशिष्ट समाधान शक्ति के साथ मास स्पेक्ट्रोमीटर का उपयोग करते समय योजक द्रव्यमान दोष के कारण मोनोआइसोटोपिक द्रव्यमान को मापा नहीं जा सकता है। इसके बजाय, नाममात्र या औसत द्रव्यमानमापा जाता है या रिपोर्ट किया जाता है 5,10,11,12,13। क्यूरेटेड द्रव्यमान14,15 के लिए उपयोग किए जाने वाले स्रोत के आधार पर औसत मौलिक द्रव्यमान भिन्न हो सकते हैं। जबकि मौलिक द्रव्यमान में अंतर छोटे हो सकते हैं, वे बड़े-अणु आणविक भार गणना के लिए महत्वपूर्ण मूल्यों को जोड़ सकते हैं। सॉफ्टवेयर अनुप्रयोग में डिफ़ॉल्ट रूप से उपयोग किए जाने वाले औसत मौलिक द्रव्यमान पूरक तालिका 1 में दिखाए गए हैं। बायोफार्मास्युटिकल अनुसंधान और विकास (आर एंड डी) क्षेत्र जैसे विनियमित वातावरण के लिए, लगातार आणविक द्रव्यमान को बनाए रखना महत्वपूर्ण है क्योंकि द्रव्यमान में परिवर्तन नियामक फाइलिंग के दौरान आणविक इकाई में परिवर्तन का संकेत दे सकता है। तात्विक द्रव्यमान के उपयोग में स्थिरता को सक्षम करने के लिए, मौलिक द्रव्यमान का एक शब्दकोश सॉफ्टवेयर टूल के साथ अल्पविराम-पृथक मान (सीएसवी) पाठ फ़ाइल के रूप में शामिल किया गया है: Element_Mass.csv (पूरक कोडिंग फ़ाइल 1)। इसी तरह, आमतौर पर एमएबी पर देखी जाने वाली ग्लाइकन रचनाओं की एक क्यूरेटेड सूची शामिल है: ग्लाइकन.csv (पूरक कोडिंग फ़ाइल 2)। दोनों फ़ाइलें एक निष्पादन योग्य अनुप्रयोग के रूप में एक ही फ़ोल्डर स्थान में सहेजी जाती हैं और उपयोगकर्ता द्वारा एक विशिष्ट मौलिक द्रव्यमान सूची या ग्लाइकन लाइब्रेरी का उपयोग करने के लिए संशोधित की जा सकती हैं।
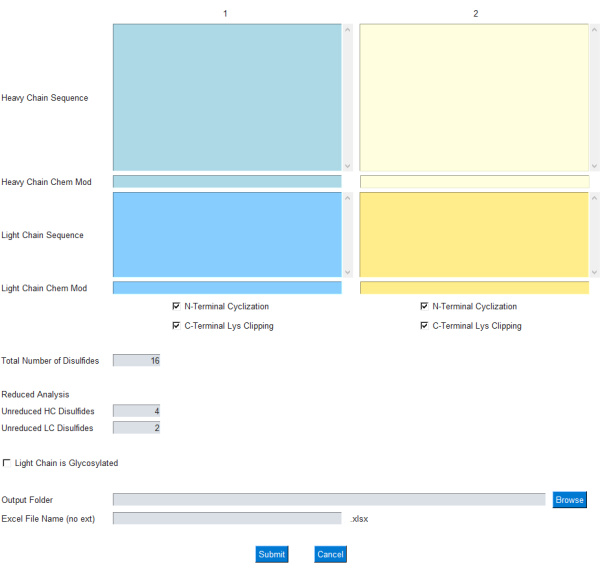
चित्र 1: mAbScale अनुप्रयोग के लिए GUI इंटरफ़ेस। जीयूआई मॉड्यूल उपयोगकर्ता को द्रव्यमान गणना के लिए इनपुट निर्दिष्ट करने की अनुमति देता है। उपयोगकर्ता हल्के और भारी एंटीबॉडी श्रृंखलाओं के लिए एकल-अक्षर अमीनो एसिड अनुक्रम दर्ज करता है। भारी-श्रृंखला एन-टर्मिनल साइक्लाइजेशन और सी-टर्मिनल लाइसिन क्लिपिंग के लिए सामान्य संशोधन चेक बॉक्स के रूप में शामिल हैं। रासायनिक सूत्र/तात्विक रचनाओं को संबंधित केम मॉड पाठ बॉक्स के माध्यम से जोड़ा/घटाया जा सकता है। कृपया इस आंकड़े का एक बड़ा संस्करण देखने के लिए यहाँ क्लिक करें.
