Son yirmi yılda, biyoterapötikler modern ilaç endüstrisinin temel dayanağı haline geldi. SARS-CoV2 salgını ve yaşamı tehdit eden diğer koşullar, biyofarmasötik moleküllerin daha hızlı ve daha geniş bir şekilde geliştirilmesine olan ihtiyacıdaha da artırmıştır 1,2,3.
Biyoterapötik moleküler ağırlık, diğer analitik tahlillerle kombinasyon halinde molekülün tanımlanması için kritik öneme sahiptir. Sağlam ve azaltılmış alt birim kütleleri, QTPP’de (Kalite Hedef Ürün Profili)4 açıklandığı gibi, kaliteyi korumayı amaçlayan kontrol stratejilerinin bir parçası olarak keşif ve geliştirme yaşam döngüleri boyunca kullanılır.
Biyofarmasötik endüstrisindeki analitik gelişme, büyük ölçüde bozulmamış kütle analizi için kütle ölçümlerine ve peptit haritalama veya çok özellikli yöntem (MAM) izleme kullanılarak derin karakterizasyona dayanır. Modern kütle spektrometresi (MS) platformlarını kullanan bu tekniklerin merkezinde, yüksek çözünürlüklü doğru kütle (HR/) ölçümleri sağlama yeteneği vardır. Çoğu HR/cihazı, kütle aralığına göre ölçeklenen 0,5-5 ppm aralığında kütle doğruluğu sağlar. Bozulmamış büyük moleküller için kütleleri doğru bir şekilde ölçme yeteneği, büyük moleküllü terapötiklerin hızlı ve güvenli bir şekilde tanımlanmasını sağlar. Büyük moleküller için tipik deney koşulları (>10 kDa) kullanılarak izotopik çözünürlük elde edilemediğinden, karşılaştırma ve tanımlamaiçin ortalama kütleler hesaplanmalıdır 5,6.
Tipik bir bozulmamış veya alt birim protein kütle spektrumu, translasyon sonrası modifikasyonlardan (PTM) kaynaklanan çeşitli moleküler formlar ve klipler veya dizi varyantları gibi herhangi bir birincil yapı farklılığı hakkında kompozit bilgiler içeren genel proteoform profilini temsil eder. Bu ölçümlerin nispeten kolay ve yüksek verimli doğası, onları karakterizasyon ve proses içi izleme kontrolleri için çekici kılmaktadır 7,8. Bu deneyler için veri analizi genellikle kullanıcının moleküler formlar (PTM’ler veya diğer moleküler formlar aralığı) için arama alanını tanımlamasını gerektirir. Glikosile edilmiş proteinler için, bu arama alanı büyük ölçüde glikoform heterojenliği tarafından yönlendirilir. Çoklu PTM’lerin, disülfür bağ konfigürasyonlarının ve birincil yapı boyunca diğer varyasyonların kombinasyonları, olası tüm moleküler formların hesaplanmasını sıkıcı bir görev haline getirir. Bu nedenle, olası moleküler formların manuel olarak hesaplanması, insan hatası potansiyeli yüksek, zaman ve kaynak tüketen bir süreçtir.
Burada, mAbs, bsAbs, ADC’ler vb. gibi biyoterapötik moleküllerin en önemli özellikleri göz önünde bulundurularak geliştirilmiş bir kütle hesaplama aracı sunuyoruz. Araç, kütlelerin ve temel bileşimlerin tutarlı bir şekilde hesaplanması için arama alanı değişkenlerinin kolayca birleştirilmesine olanak tanır. Bu aracın modüler yapısı, daha da geliştirilmesini ve diğer modaliteler için kütle hesaplaması ve kütle eşleştirmesine uygulanmasını sağlayacaktır.
GUI modülü, kullanıcının Şekil 1’de gösterildiği gibi kütle hesaplaması için girişi belirlemesine izin verir; Spesifik olarak, kullanıcı hafif ve ağır antikor zincirleri için tek harfli amino asit dizileri girer. Ağır zincirli N-terminal siklizasyonu ve C-terminal lizin kırpma için yaygın değişiklikler onay kutuları olarak dahil edilmiştir. Ayrıca, kimyasal formül/element bileşimi, ilgili Chem Mod metin kutusu aracılığıyla bu protein zincirlerine eklenebilir/çıkarılabilir. Bu, kullanıcıya, bir ADC durumunda birden fazla translasyon sonrası modifikasyon veya küçük moleküllü bir yük içeren bir temel bileşim ekleme esnekliği sağlar. Çoğu terapötik mAb, hafif zincirdeki glikozilasyon bölgelerini uzaklaştırmak için tasarlandığından, hafif zincirdeki glikozilasyon isteğe bağlı bırakılır ve GUI’deki bir onay kutusu kullanılarak belirtilebilir.
Antikorlar için bozulmamış kütle analizinin tipik bir varyasyonu, zincirler arası disülfür bağlarını azaltarak hafif zincirin ağır zincirden ayrıldığı indirgenmiş bir alt birim kütle analizidir. Kullanılan indirgeyici ajanın gücüne bağlı olarak, zincir içi disülfür bağları parçalanabilir veya bölünmeyebilir. Kullanıcılar, IgG alt tipine bağlı olarak veya sistein konjuge ADC9 durumunda toplam disülfür bağı sayısını girme esnekliğine sahiptir.
Uygulama, kütleleri aşağıdan yukarıya doğru hesaplar, burada temel bileşimler ilk olarak bireysel ağır zincirler ve hafif zincirler için hesaplanır. Daha sonra, ağır zincir (HC) N-terminal siklizasyonu Lys-clipping, hesaplanan temel bileşimlerin ayarlanmasıyla hesaba katılır. Belirtilen herhangi bir kimyasal modifikasyon daha sonra ağır ve/veya hafif zincirlere uygulanır. Analizin türüne ve kullanıcı tarafından belirtilen disülfür-bağ modellerine bağlı olarak, iki polipeptit zinciri için hidrojen sayısı ayarlanır. Glikosile HC ve hafif zincir (LC) (isteğe bağlı) kütleleri, kullanıcının girdisine göre hesaplanır. Son olarak, birden fazla HC ve LC kütlesi birleştirilir ve bozulmamış kütle hesaplaması için disülfür bağ numaraları otomatik olarak güncellenir.
Bozulmamış proteinler gibi daha büyük moleküllerde, tipik çözme gücüne sahip kütle spektrometreleri kullanılırken ilave kütle kusuru nedeniyle monoizotopik kütleler ölçülemez. Bunun yerine, nominal veya ortalama kütlelerölçülür veya rapor edilir 5,10,11,12,13. Ortalama temel kütleler, küratörlüğünde kütleler14,15 için kullanılan kaynağa göre değişebilir. Element kütlelerindeki farklılıklar küçük olsa da, büyük moleküllü moleküler ağırlık hesaplamaları için önemli değerler ekleyebilirler. Yazılım uygulamasında varsayılan olarak kullanılan ortalama element kütleleri Ek Tablo 1’de gösterilmektedir. Biyofarmasötik araştırma ve geliştirme (Ar-Ge) alanı gibi düzenlenmiş ortamlar için, tutarlı moleküler kütlelerin korunması önemlidir, çünkü kütlelerdeki değişiklikler, düzenleyici başvurular sırasında moleküler varlıkta değişiklikler anlamına gelebilir. Element kütlelerinin kullanımında tutarlılığı sağlamak için, yazılım aracına virgülle ayrılmış değer (csv) metin dosyası olarak bir element kütleleri sözlüğü eklenmiştir: Element_Mass.csv (Ek Kodlama Dosyası 1). Benzer şekilde, mAb’lerde tipik olarak görülen glikan bileşimlerinin küratörlüğünde bir listesi dahil edilmiştir: Glikan.csv (Ek Kodlama Dosyası 2). Her iki dosya da çalıştırılabilir bir uygulamayla aynı klasör konumuna kaydedilir ve kullanıcı tarafından belirli bir temel kütle listesi veya glikan kitaplığı kullanacak şekilde değiştirilebilir.
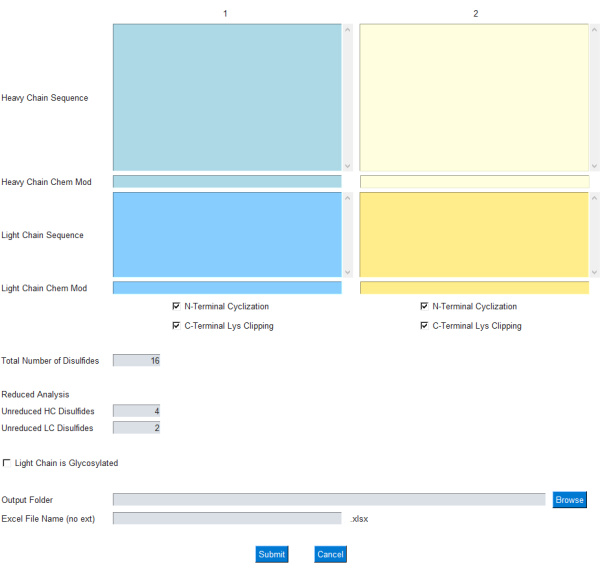
Şekil 1: mAbScale uygulaması için GUI arayüzü. GUI modülü, kullanıcının kütle hesaplaması için girişi belirlemesine izin verir. Kullanıcı, hafif ve ağır antikor zincirleri için tek harfli amino asit dizileri girer. Ağır zincirli N-terminal siklizasyonu ve C-terminal lizin kırpması için yaygın değişiklikler onay kutuları olarak dahil edilmiştir. Kimyasal formüller/temel bileşimler, ilgili Chem Mod metin kutusu aracılığıyla eklenebilir/çıkarılabilir. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
