We performed an experiment to determine the transcriptional induction of the antimicrobial peptide gene Drosocin, following injury and bacterial infection of Drosophila. We compared the levels of Drosocin expression under three conditions, injection of PBS (injury), injection of E. coli (bacterial infection), and uninjured control. Samples of whole flies were collected 6 hours after the challenge, typical for an infection experiment in innate immunity research. The experiment was performed in three biological replicates. The original reads of the first biological replicate from the RT-qPCR from the instrument, and log representation, are shown in Figure 1. Figure 1A shows the direct fluorescence signal, while the graph in Figure 1B shows the log representation of the fluorescence intensity. The threshold value used to calculate the Cq of the samples is set at a fluorescence intensity of 0.4 (Fig. 1B). The Cq values for all replicates were used to calculate the ΔCq and ΔΔCq as explained above, and summarized in Table 2. We performed the same calculations for three replicate experiments, determining ∆∆Cq values and the fold RNA changes (Table 2). Ultimately, we calculated the mean and SD of the log2 fold changes among the three biological replicates for each experimental condition (Table 2), and plotted the results in a bar chart in Figure 2. The results represent fold changes in Drosocin RNA concentration after injection, compared to uninjected flies.
Drosocin is an antimicrobial peptide (AMP), produced as part of the innate immune response to infection (Tzou et al., 2000). Our measurements confirm a strong induction of Drosocin after infecting flies with E. coli (Fig. 1, 2). The innate immune system is also activated when flies are wounded, as is observed when flies are injured and injected with PBS (Lemaitre and Hoffmann, 2007) (Table 2, Fig. 2). Injury typically triggers a lower, more transient innate immune response than bacterial infection (Lemaitre and Hoffmann, 2007). This is indeed observed in our analysis, where injury (injection of PBS) triggers a 10-fold increase in Drosocin expression, while flies infected with E. coli boost the production of Drosocin 30 times over the non-treated flies (Table 2, Fig.2).
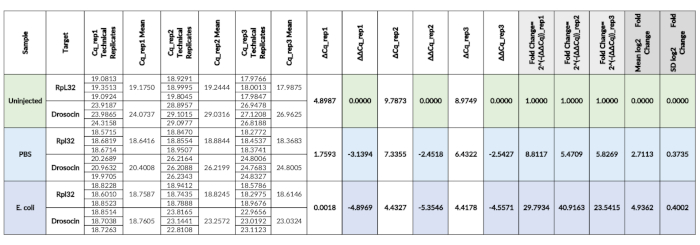
Table 2. qPCR calculations. Cq values of samples in three biological replicates (rep1, -2, -3), each in technical triplicates, obtained directly from the ViiA 7 Real-Time PCR system. Cq means (columns D, F, H) are determined as the mean of the three technical replicates of each biological replicate sample. From the Cq mean values, ΔCq values (columns I, K, M) and ΔΔCq values (columns J, L, N) are calculated for each biological replicate. ΔΔCq values are subsequently converted into fold changes (columns O, P, Q). For the final display of the data, the mean and SD of the data is determined, here presented in log2 scale (columns R, S). Please click here to view a larger version of this table.
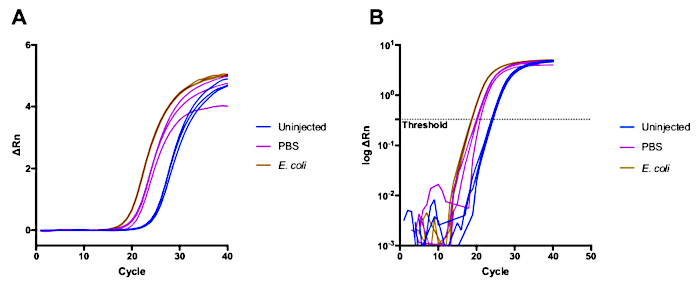
Figure 1. Fluorescent intensity represented as ΔRn. The value for ΔRn is obtained by subtracting the baseline fluorescence (Rn-) to the measured fluorescence (Rn+). In (A) ΔRn is plotted in a linear scale, while in (B) it is plotted using a log scale. The slashed line represents the threshold used to calculate the Cq of each sample. Please click here to view a larger version of this figure.
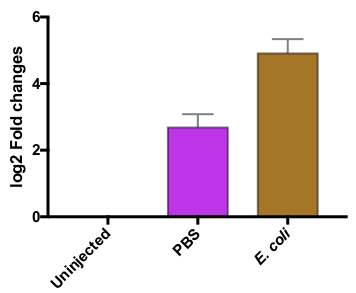
Figure 2. Changes in Drosocin RNA expression following injury and bacterial infection. Log2 transform of the fold RNA changes are shown. Flies injected with sterile PBS show a log2 fold increase of 2.7 in Drosocin expression, while flies infected with E. coli boost Drosocin expression by a log2 fold change of 4.9, compared to uninjected flies. Error bars represent the SD of the 3 biological replicates.