An In Vitro Assay to Quantify the Intracellular Growth of Parasite in Macrophages
Abstract
Source: Sarkar, A., et al. Quantification of Intracellular Growth Inside Macrophages is a Fast and Reliable Method for Assessing the Virulence of Leishmania Parasites. J. Vis. Exp. (2018).
This video demonstrates an assay to quantify the intracellular growth of parasites in bone marrow-derived macrophages (BMMs). The macrophage cells are infected with the motile and non-motile invasive stages of Leishmania, metacyclic promastigotes and amastigotes, respectively. The parasites invade the macrophages, getting internalized into a parasitophorous vacuole and multiplying inside. The intracellular parasite growth is quantified using a microscope upon fluorescently staining the macrophage and parasite nuclei.
Protocol
1. Plating BMMs on Coverslips for Infection
- Prepare 6-well tissue culture plates by placing four (4) sterile 12 mm glass coverslips (autoclaved and stored in a closed glass container) in the bottom of each empty well.
NOTE: Pick each sterile 12 mm coverslip up with an aspirating tip fitted to a vacuum line. Place coverslips into the wells without overlap, releasing them at the desired position by pinching the tube to interrupt the vacuum. - Aspirate media (and any non-adherent cells) from Petri dishes coated with adherent bone marrow-derived macrophages and gently rinse adherent cells 2x with sterile Dulbecco's Phosphate buffered saline without calcium and magnesium chloride (PBS -/-) pre-warmed to 37 °C.
- Add dropwise, 5 mL sterile 1x PBS -/- with a final concentration of 1 mM EDTA to each Petri dish and incubate for 5 min at 37 °C to detach adherent BMMs. Verify that the cells are detached using a microscope.
- Collect all detached cells suspended in 1x PBS -/- supplemented with 1 mM ethylenediaminetetraacetic acid (EDTA) in a 50 mL conical tube. Rinse each dish 2x with 5 mL PBS -/- and add to the cell suspension pool.
NOTE: Cell suspension may be diluted with additional PBS -/- to protect macrophages from EDTA toxicity during the collection process. - Centrifuge at 300 x g for 10 min at 4 °C. Discard the supernatant and resuspend macrophages in 5 – 10 mL BMM medium by pipetting. Place resuspended cells on ice to prevent attachment to tubes and clumping.
- Count viable cells using Trypan blue exclusion in the hemocytometer.
NOTE: To count BMM cells resuspended in 5 mL medium, a dilution mix of 25 µL cell suspension in 445 µL PBS -/- and 30 µL 0.4% Trypan blue solution typically allows easy quantification in a hemocytometer (taking into account a 20x dilution factor for the final calculation). Count only cells that did not get stained with Trypan blue (viable cells). The volume of cell suspension may be altered if this mixture is too concentrated or diluted. In general, the yield of BMM per preparation varies between 2 x 107 and 5 x 107 cells from a single mouse. - Prepare the cell suspension in BMM medium at desired plating concentration (5 x 105/mL) and disperse suspension to 6 well plates in 2 mL medium per well so that each well receives 1 x 106 macrophages.
NOTE: This is a critical step that ensures every well gets the adequate number of macrophages to cover the coverslips uniformly. Check that coverslips in wells are not overlapping or floating in the medium. If so, gently adjust with a sterile pipette tip. - Incubate overnight at 37 °C, 5% CO2.
2. Purification of Infective Forms of L. amazonensis
NOTE: Prepare Leishmania for infections- purify metacyclic promastigotes from stationary promastigote cultures or differentiate promastigotes in culture into amastigote form using standard L. amazonensis axenic differentiation protocol.
- Purification of Leishmania metacyclic promastigotes using density gradient media (Materials)
- Prepare 40% stock solution of density gradient media in sterile, endotoxin-free water.
- Dilute density gradient solution in 10x M199 medium (without serum) to prepare 10% concentration in the M199 medium.
- Filter all solutions through 0.22 µm filter.
NOTE: Stock solutions can be stored at 4 °C in darkness for no longer than 1 month. - Add 2 mL of 40% density gradient media solution in the bottom of a 15-mL conical centrifuge tube.
- Layer 2 mL of 10% density gradient media solution in M199 on top of the 40% density gradient media layer carefully, using a Pasteur pipette to avoid any mixing between the two layers.
- Collect 1 x 109 parasites from stationary-phase culture by centrifugation at 1,900 x g for 10 min.
- Resuspend cells in 6 mL Dulbecco's Modified Essential Medium (DMEM).
- Layer 2 mL of the cell suspension directly on top of the 10% density gradient media layer, gently, using a Pasteur pipette to avoid mixing between layers.
- Centrifuge the gradient for 10 min at 1,300 x g at room temperature.
NOTE: Due to differences in physical properties between Leishmania species, centrifugation conditions for each might have to be slightly adjusted to ensure maximum yield. - Collect parasites (enriched in metacyclic promastigote form) from the band formed at the upper 10% density gradient media boundary (interface between the 0% and 10% density gradient media layers).
- Dilute parasites with one volume of DMEM and collect by centrifugation (1,900 x g for 10 min at room temperature).
- Resuspend in 500 µL DMEM medium and count in a hemocytometer to quantify yield.
- Generation of L. amazonensis amastigotes under axenic culture conditions (low pH / elevated temperature).
- Mix 5 mL of log-phase promastigote culture (pH 7.4 at 26 oC) with an equal volume of amastigote medium (pH 4.5), using 25 cm2 flasks (total 10 mL medium) and incubate at 26 °C overnight.
- Shift the flask from 26 °C to 32 °C.
- After 3 or 4 days, split culture 1:5 in amastigote media at 32 °C.
- Check the parasites in the next 3 – 4 days (maximum 7 days) to see if they are ready for use in infections.
NOTE: Healthy axenic amastigotes should have an oval shape, without visible flagella. Partially differentiated amastigotes have a large oval shape with short flagella. The culture should not have a lot of clumps – many clumps are an indication of non-growing, dying parasites. The amastigote culture can be split 1:5 and maintained for a maximum of 3 weeks.
3. Infection with L. amazonensis
- Dilute parasite suspensions according to desired MOI (usually 3-5 metacyclic promastigotes per macrophage [MOI of 1:3 or 1:5] and 1 amastigote per macrophage [MOI 1:1]). Add Leishmania in PBS -/- in a volume of 50 – 100 µL to each well already containing 2 mL medium.
- Incubate BMMs for 1 h with amastigotes and 3 h with metacyclic promastigotes at 34 oC for infection.
NOTE: Optimal infection temperature may vary by species. - Following incubation, thoroughly wash away free parasites in each well 3x with 2 mL PBS -/- pre-warmed to 37 °C.
NOTE: Disperse PBS gently to ensure that coverslips do not float over each other. Gently swirl the plate and then aspirate out the liquid. Use separate aspirating tips if using multiple strains of parasites to avoid cross-contamination. - Fix 1 h or 3 h time-point samples by incubating each well with 1.5 – 2 mL of 2% PFA in PBS -/- for 10 min. Wash 3x with PBS -/-. Do not aspirate last wash and refrigerate plates containing coverslips in PBS until staining.
- Add 2 mL fresh BMM medium to each well on plates for further time-points and incubate at 34 °C.
- Fix remaining time-points as desired as described in step 4.3, and refrigerate plates until staining.
4. DAPI Staining and Coverslip Mounting
- Aspirate PBS from wells containing coverslips and add 1.5 mL PBS -/- with 0.1% non-ionic detergent. Incubate for 10 min at room temperature, followed by 3x wash with 2 mL PBS -/-.
- Add 1 mL PBS -/- with 2 µg/mL 4′,6-diamidino-2-phenylindole (DAPI) (diluted from 5 mg/mL stock solution in water) to wells containing coverslips and incubate for another 1 h at room temperature.
NOTE: The incubation time with DAPI is critical for proper visualization of intracellular Leishmania parasites. Macrophage nuclei stain rapidly, due to their larger size and DNA content but staining of nuclei of the intracellular parasites is slower. Thus, extending incubation time beyond what is required to visualize the BMM nuclei is necessary to allow DAPI to permeate through and the parasite plasma membrane, and the parasite nuclear membrane. With proper DAPI stain, the mitochondrial kinetoplast DNA near the flagellar pocket of the parasite can also be visualized, in addition to the nuclear DNA. - Wash each well containing coverslips 3x with 2 mL PBS -/- and then lift and flip coverslips with forceps to place the cell side down and mount on glass microscope slides with a commercially available antifade mounting reagent.
NOTE: Coverslips are extremely fragile and need cautious handling. Avoid putting pressure on them, especially against the sidewall of the wells when lifting. Adding a few drops of PBS -/- reduces the surface tension between the coverslip and the well and facilitates the lifting process. A fine gauge needle may be used to assist in prying the coverslips from the bottom of the well. After placing a coverslip on the slide, gently press down with forceps to push out any air bubbles in the mounting media. If a coverslip is broken or has possibly flipped during the lifting process, do not remount; simply use the spare fourth coverslip. - Refrigerate slides until quantification.
5. Infection Quantification
- Examine DAPI-stained slides under a fluorescence microscope by focusing on macrophages using a 100X objective lens with immersion oil (excitation at 358 nm; optimal emission at 461 nm).
Ensure that the focus is on the layer of macrophages between the glass slide and coverslip. - Quantify the number of macrophages (large nuclei stained with DAPI) and the number of smaller amastigote nuclei clustered around each macrophage nucleus (see Figure 1A, DAPI) for each field of vision, using a manual counter.
NOTE: Amastigote nuclei may not all be visible in one plane of focus due to their small size. Count those that are visible when focused on macrophage nuclei, and then use fine focus to check for parasite nuclei in planes above and below the macrophage nuclei. - Move to another field of vision to repeat quantification. Use a separate counter key to track the number of fields counted. Move through visual fields in parallel rows across each coverslip, to prevent counting overlap.
- Quantify a minimum of 200 macrophages per coverslip. Quantify each time-point of infection in triplicate (3 coverslips). Count the fourth coverslip if one of the previous ones is not suitable for counting due to coverslip overlap, poor mounting, glass breakage, etc.
NOTE: If 200 macrophages cannot be counted on any one coverslip, count the spare fourth. - Calculate infection rates as amastigotes/macrophage or amastigotes/100 macrophages and determine the percent infected macrophages from the raw quantification data.
Representative Results
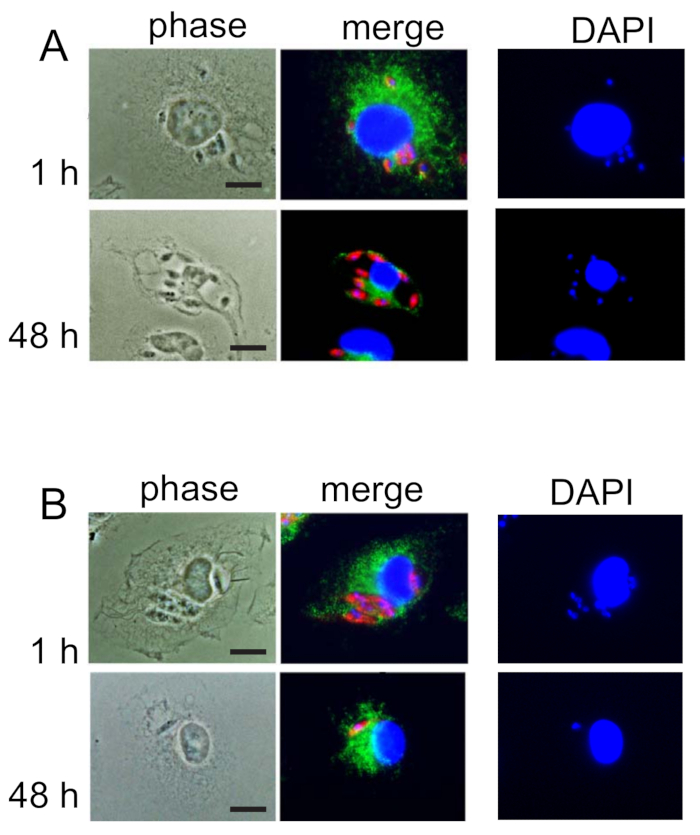
Figure 1: A representative illustration of BMM infection with virulent axenic amastigotes (A) and non-virulent log-phase promastigotes (B) of L. amazonensis. Immunofluorescence images of macrophages isolated from BALB/c mice, 1 h and 48 h following infection. Infected macrophages were processed for immunofluorescence as described in the protocol. PV membranes were stained with rat anti-mouse Lamp1 monoclonal antibody (1:1,000 dilution) for 1 h, followed by 1 h incubation with anti-rabbit fluorescent IgG (1:500 dilution). Parasite staining was performed by incubating coverslips with mouse polyclonal antibodies generated against axenic L. amazonensis amastigotes, followed by anti-mouse IgG red dye (1:500 dilution). All coverslips were further treated with DAPI for staining nuclei. (A) Formation of distinct PVs harboring multiple amastigotes at 48 h time point is characteristic of a successful Leishmania infection with axenic amastigotes. (B) The absence of distinct PV formation and replicating amastigotes at a 48 h time point typifies a lack of virulence in promastigotes from log-phase culture. Red indicates anti-Leishmania staining, green indicates anti-Lamp1, blue indicates DAPI-stained DNA, and yellow indicates a merge of anti-Lamp1 and DAPI stains. Bars, 5µm. This figure has been modified from Mittra, B. et al., 2013. Originally published in J. Exp. Med.
Disclosures
The authors have nothing to disclose.
Materials
| 6 well cell culture plate | Cellstar | 657160 | |
| Adenine | Acros Organics | AC147440250 | |
| Aerosol Barrier Pipet Tips (100-1000 μL) | Fisherbrand | 02-707-404 | |
| Aerosol Barrier Pipet Tips (20-200 μL) | Fisherbrand | 02-707-430 | |
| Aerosol Barrier Pipet Tips (2-20 μL) | Fisherbrand | 02-707-432 | |
| Bard-Parker Rib-Back Carbon Steel Surgical Blade #10 | Aspen Surgical | 371110 | |
| BD Luer-Lok Tip 10 mL Syringe | Becton Dickinson (BD) | 309604 | |
| BD Precisionglide Needle, 25G | Becton Dickinson (BD) | 305124 | |
| Cell Culture Dish 35 mm x 10 mm | Cellstar | 627 160 | |
| Cell Culture Flask | Cellstar | 660175 | |
| Cover Glasses: 12 mm circles | Fisherbrand | 12-545-80 | |
| DAPI (4',6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | D1306 | |
| D-Biotin | J.T. Baker | C272-00 | |
| EDTA | Sigma Aldrich | EDS | |
| Ethyl alcohol 200 proof | Pharmco-AAPER | 111000200 | |
| Falcon 100 mm x 15 mm non-TC-treated polystyrene Petri dish | Corning | 351029 | |
| Fetal Bovine Serum | Seradigm | 1500-500 | |
| Ficoll400 | Sigma Aldrich | F8016 | |
| Fluorescence Microscope | Nikon | E200 | |
| Goat anti-mouse IgG Texas red | Invitrogen | T-862 | |
| Goat anti-rabbit IgG AlexaFluor488 | Invitrogen | A-11034 | |
| Hemin | Tokyo Chemical Industry Co. LTD | H0008 | |
| HEPES (1M) | Gibco | 15630-080 | |
| Isoton II Diluent | Beckman Coulter | 8546719 | |
| L-Glutamine | Gemini | 400-106 | |
| Medium 199 (10X) | Gibco | 11825-015 | |
| Na pyruvate (100 mM) | Gibco | 11360-070 | |
| Paraformaldehyde | Alfa Aesar | 43368 | |
| Penicillin/Streptomycin | Gemini | 400-109 | |
| Phosphate Buffered Saline (-/-) | ThermoFisher | 14200166 | |
| Polypropyline conical Centrifuge Tubes 15 mL | Cellstar | 188 271 | |
| Polypropyline conical Centrifuge Tubes 50 mL | Cellstar | 227 261 | |
| ProLong Gold antifade reagent | ThermoFisher | P36930 | |
| Rat anti-mouse Lamp-1 antibody | Developmental Studies Hybridoma Bank | 1D4B | |
| Recombinant Human M-CSF | PeproTech | 300-25 | |
| Reichert Bright-Line Hemocytometer | Hausser Scientific | 1492 | |
| RPMI Medium 1640 (1X) | Gibco | 11875-093 | |
| Triton X-100 Surfactant | Millipore Sigma | TX1568-1 | |
| Trypan Blue | Sigma Aldrich | T8154 | |
| Delicate Scissors, 4 1/2" | VWR | 82027-582 | |
| Dissecting Forceps, Fine Tip | VWR | 82027-386 | |
| Microscope Slides | VWR | 16004-368 | |
| Z1 Coulter Particle Counter, Dual Threshold | Beckman Coulter | 6605699 |
