1. Poly-D-Lysine (PDL): Preparation
- Add 5 ml of sterile ddH2O to 5 mg of PDL to obtain a stock solution of 1 mg/ml.
- Mix stock solution by pipetting several times.
- Use immediately or store Poly-D-Lysine Solution at 2-8 °C.
2. Poly-D-Lysine (PDL): Coating Plastic Cell Culture Dishes
- Dilute the PDL stock solution with sterile ddH2O to the final concentration of 10 μg/ml.
- Pipette enough solution into a 60 mm dish to cover culture surface area (3 ml for a 60 mm dish).
- Rock gently to ensure even coating of the culture surface.
- Incubate coated plates at room temperature (RT) overnight.
- On the next day, usually the day of dissection, remove the Poly-D-Lysine Solution by aspiration and wash briefly with 3 ml of sterile ddH2O. Repeat this step. After the second wash, remove water completely by aspiration.
- Plates can be stored at 4 °C for up to three weeks.
3. Poly-D-Lysine (PDL) and Laminin: Preparation and Coating of Glass Two-chamber Slides
- Mix PDL (1 mg/ml) and laminin (1 mg/ml) stock solutions in sterile ddH2O to the final concentration of 10 and 5 μg/ml, respectively.
- Pipette enough solution into wells of a glass two-chamber slide to cover the culture surface area (1 ml for each well of a 2 well glass two-chamber slide).
- Rock gently to ensure even coating of the culture surface.
- Incubate coated plates at RT overnight.
- On the next day, remove the Poly-D-Lysine-Laminin coating solution via aspiration and wash briefly twice with 1 ml of sterile ddH2O. After the second wash, remove water completely by aspiration.
- Chamber slides can be stored at 4 °C for up to three weeks.
Note: Any glass chamber slide can be coated following this protocol. We often use the two-chamber slides because each slide provides the control-test experimental setting (e.g. untreated versus treated, untransfected versus transfected).
4. Neuronal Dissection and Culture
- Warm the following reagents in a 37 °C water bath:
- TrypLE Express on its original 100 ml bottle.
- Neurobasal/B27 complete medium (see table I). The volume warmed depends on the number of dishes to be plated (e.g. 30 ml for ten 60 mm plated dishes).
- Add 3 ml of cold Hibernate E solution to four 60 mm culture dishes and 13 ml to a 15 ml BD Falcon high clarity polypropylene conical tube.
- Add 25-30 ml of cold dissection medium (see table II, Dr. Olimpia Meucci, personal communication) to each of three 100 mm culture dishes. These plates, containing a large volume of medium, will be used to wash the embryos immediately after their removal from the amniotic sacs (steps 4.7 and 4.8).
- Euthanize an E17 timed pregnant rat by CO2 in accordance with Public Health Services Policy on Humane Care and Use of Laboratory Animals and under an institutionally approved animal care and use protocol.
- Spray lower abdomen with 70% EtOH and cut medially through the skin and muscles with a pair of scissors exposing the uterus and embryos.
- Remove all fetuses and place them in a sterile 100-mm dish containing an excess of cold dissection medium (25-30 ml, see step 4.3).
- Cut embryos using a small pair of scissors from amniotic sac and place them in the second 100-mm dish containing cold dissection medium.
- Wash the embryos at room temperature by gently tilting the 100-mm dish for 5-10 seconds. Then, transfer the rinsed embryos to the third 100 mm dish containing dissecting medium. Two washes in excess medium are usually sufficient to remove all traces of blood. However, if necessary, wash one more time using a fresh 100-mm dish containing 25-30 ml of cold dissection medium.
- Using a stereomicroscope and curved forceps, extract each rat embryo’s brain by pulling out the skin and skull. Place entire brain in one of the 60-mm dishes (typically, with no more than 5 brains per dish) with cold Hibernate E. Keep these plates on ice.
- Take one dish at a time and, under a dissecting microscope, separate the hemispheres and isolate the cerebral cortices removing the midbrain and meninges.
- Optional: cut brains along midline, extract hippocampi, and follow the procedure below to isolate hippocampal neurons.
- Collect all the dissected cortices in a 15-ml clear conical tube containing 13 ml of cold Hibernate E. Leave the cerebral cortices on ice until all the dissections are completed. Due to their small size, dissected hippocampi can be collected in a 1.5-ml Eppendorf tube instead of a 15-ml tube. If desired, at this step cortices or hippocampi can be placed in a cryotube vial containing 1 ml of Hibernate E + 2% B27+ Gentamicin (50 μg/ml) + Fungizone (250 ng/ml) in the proportion of 2-4 cortices or 2-4 hippocampi per vial. Brain tissue can be stored at 4 °C in the dark for up to one week (later times have not being tested yet). When needed, use fine forceps to transfer the brain tissue to a 15 ml tube containing Hibernate E and then follow the protocol below to isolate neurons.
- Transfer the tube to a tissue culture hood. Allow the cortices to settle to the bottom of the tube and then carefully remove the supernatant.
- Add 13 ml of fresh Hibernate E to the 15-ml conical tube, allow cortices settle at the bottom of the tube and carefully remove the supernatant. Repeat this step 2 more times and, after the last wash, carefully remove all media.
- Enzymatically digest cerebral cortices by adding 1-2 ml (depending on the number of cortices; use less for hippocampi isolation) of warm TrypLE Express. Seal the cap of the tube with Parafilm and float the tube in a 37 °C water bath for 10 minutes.
- Spray the tube with 70% Ethanol before opening the cap and add 10 ml of Hibernate E. Allow the cortices to settle at the bottom of the tube and remove the supernatant. Repeat this step three times to wash out TrypLE Express. In last step, carefully remove all media.
- Gently triturate (4-5 times) the cortices in 2 ml of Neurobasal/B27 complete medium using a fire-polished glass Pasteur (approximately 1 mm in diameter). Be careful to avoid bubbles.
- Repeat another 4-5 times with a sterile glass Pasteur pipette smaller in diameter (i.e. a pipette about 1/2-3/4 mm in diameter). Do not use a Pasteur pipette smaller than this or it will destroy the cells.
- Allow remaining pieces of tissue (generally very few, if any) to settle.
- Transfer the upper single-cell suspension to a new 15-ml tube, leaving behind settled pieces of tissue. Further dilute the cell suspension up to 10-12 ml with Neurobasal/B27 complete medium.
- Mix well and dilute cells for counting by adding 10 μl of cell suspension to 490 μl of 50x Counting Solution (see table III) in a 1.5 ml Eppendorf tube.
- Plate cells on PDL-coated plates at the density of 5.0 x 104/cm2. If nucleofection is to be performed, we recommend plating the cells at a higher concentration (8-10 x 104/cm2).
- Normally, we dissect 9-10 fetuses per experiment, as approximately 13 x 106 neurons are derived from each E17 fetus. If more embryos are needed, make sure that the whole procedure does not last more than two hours.
- If desired, 24 hours after isolation, 10 μM of cytosine-β-D-arabinofuranoside (AraC) can be added to each dish, in order to prevent glial proliferation. However, this step is not required since Neurobasal/B27 medium inhibits glial proliferation, as per manufacturer’s recommendations (Invitrogen/Gibco).
- Neurons can be used for experiments after 4-5 days in vitro, although exact time depends on desired differentiation stage. We have cultured neurons for up to 4 weeks without a significant decrease in survival (Figure 1).
- For extended culturing, replace culturing medium every week with freshly prepared Neurobasal/B27 complete medium.
5. Representative Results
Neurons cultured on glass chamber slides can be subjected to immunocytochemistry. Figure 1 shows a typical image of a cortical neuron fixed after five days in culture and immunolabeled with anti-MAP-2 antibody to show neuronal processes.
Figure 2 shows a representative image of a rat hippocampal neuron after 3 weeks in culture. The neuronal morphology of a fully differentiated cell is highlighted by MAP-2 immunolabeling (MAP-2 neuronal marker, mouse monoclonal antibody clone AP-20, Gene Tex, Irvine, CA), following a standard procedure as previously described1. The images were visualized with the Nikon Eclipse E400 upright fluorescence microscope equipped with EXI aqua camera (Qimaging), motorized Z-axis, and SlideBook5 acquisition/deconvolution software (Intelligent Imaging Innovations, Inc., Denver, CO). A series of three-dimensional images of each individual picture were deconvoluted to one two-dimensional picture and resolved by adjusting the signal cut-off to near maximal intensity to increase resolution.
Figure 3 shows purity of neuronal cultures. Protein lysates were obtained from DIV7 rat neuronal cultures (ctx) and from a case of human glioblastoma (GBM). As expected, the neuronal lysate is strongly positive for the neuronal protein MAP-2 and negative for the astrocytic marker GFAP; while the GBM protein lysate is negative for MAP-2 and positive for GFAP.
Although in our protocol we have been using Hibernate E for several years as dissecting and rinsing medium, recently we have explored an additional and very practical use of it to preserve brain tissues for further use. Figure 4 illustrates a days in vitro 5 (DIV5) culture of rat cortical neurons isolated from cortices kept at 4 °C for one week in Hibernate E + B27 after their original dissection from the embryos. Neurons were plated on a glass two-chamber slide coated with PDL and laminin as previously described. The acquired image was deconvoluted using SlideBook5 acquisition/deconvolution software as described above (Figure 2).
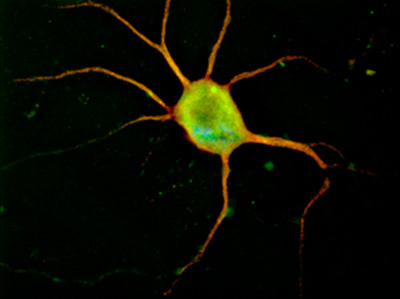
Figure 1. Representative image of a cortical neuron nucleofected with pmaxGFP (Amaxa, Lonza, Walkersville, MD) and immunolabeled with MAP-2 antibody, in red. Original magnification 100x.
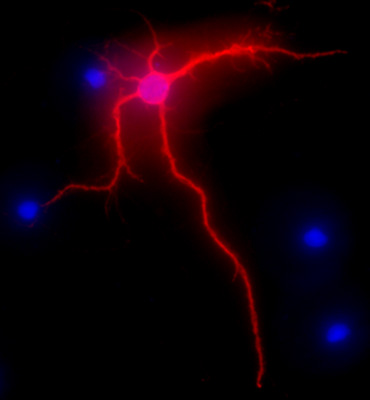
Figure 2. Representative image showing MAP-2 immunolabeling, in red, of hippocampal neurons after 3 weeks in culture. DAPI staining, in blue, shows cellular nuclei. Original magnification 40x.
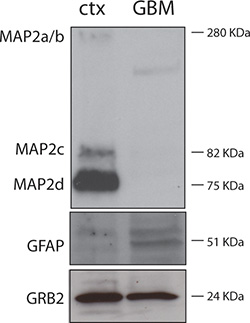
Figure 3. Western blot showing purity of neuronal cell cultures. 30 μg of rat neuronal and human GBM protein lysates were separated by electrophoresis and subjected to Western blot analysis following standard procedures1. Anti-MAP-2 was a rabbit polyclonal from Cell Signaling (Danvers, MA), anti-GFAP antibody was a mouse monoclonal from Chemicon (Millipore, Billerica, MA), and the mouse monoclonal anti-GRB2 antibody was from BD Transduction Laboratories (Sparks, MD). GRB2 was used as a loading control.
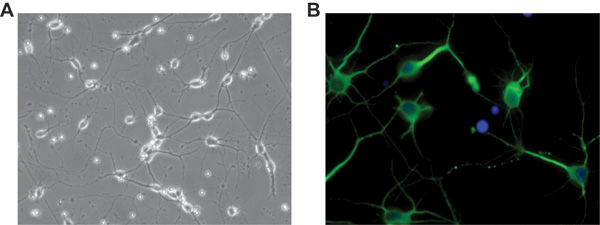
Figure 4. Representative pictures of days in vitro 5 (DIV5) rat cortical neurons obtained from cortices left in Hibernate E + B27 at 4 °C for one week after their dissection. A) Phase contrast of neurons cultured on a glass two-chamber slide. Original magnification 20X. B) Immunofluorescence showing expression of MAP-2 in neuronal processes, in green; the culture was negative for the astrocytic marker GFAP. DAPI staining, in blue, indicates cellular nuclei. Original magnification 40x.
