蛋白和蛋白复合物的晶体结构的数量近年来迅速增加。他们提出这些蛋白质的结构组织的非常宝贵的快照,并为结构 – 功能分析的基础。然而,蛋白质和构象变化,这对于其功能必需的动力,也很少由X-射线结晶学揭示。冷冻电镜,另一方面,能够捕获蛋白和蛋白复合物在不同的构象,但一般不能解析的构象变化下降到二级结构的第1级。在原子细节的蛋白质在溶液中的构象动力学只能通过NMR来解决,但这种方法仍局限于相对较小的尺寸(通常≤30 kDa)的蛋白,并且需要高浓度的蛋白质(≥100微米),这阻碍了实验低聚或易聚集的蛋白质2。一种方法,该方法能够高分辨率X-射线晶体学和低温电镜和其之间的桥梁不是由蛋白质大小或浓度的限制是酰胺氢-1与质谱(MS)结合的H / 2 H-交换(HX)。近年来,这种方法已经发展到用于蛋白质动力学,蛋白质折叠,蛋白质稳定性和构象变化3-5的分析有价值的分析工具。该方法的分子基础是骨干酰胺氢中的蛋白质,这将与氘原子交换,当蛋白质被放置在D 2 O中的溶液中不稳定的性质。随着时间的推移随后增加蛋白质量的测量与高分辨率质谱。
总之非结构化肽HX只依赖于温度,催化剂浓度(OH – ,H 3 O +,即 pH值, 见图3)和相邻的残基,由于电感,猫氨基酸侧链alytic和空间位阻效应。在固有的化学交换率k CH这些影响已经由白等人 。6被量化的优雅和程序是可用的(礼貌Z.张),其中计算K 通道的多肽取决于pH值和温度范围内每个氨基酸。在中性pH值和温度,Ķch是在10 1 -10 3秒-1的顺序。在折叠的蛋白质HX可以是2-9个数量级慢,主要是由于在二级结构的氢键和一个较小的程度,由于水合OH的受限制的访问–离子以紧密折叠的蛋白质的内部。因此HX的天然蛋白质牵连局部或全局展开,化学交换,并根据公式折叠到天然态(1)和观察到的外汇报价K OBS依赖于开放率k 运算 ,收盘率k cl和内在化学交换RA特ķ 沟道根据等式(2)。
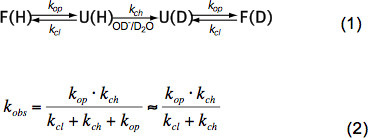
根据天然状态条件Ķ 运算比k 通道小得多,并且可以在分母被忽略。有所谓的EX1和EX2两个极端的汇率制度。如果第k CL比k 通道 (EX1)小得多所观察到的速率几乎等于开口率和HX允许直接观察的结构元件的展开的。这种汇率制度,所有的酰胺质子交换一次开盘时的结构元素,是很容易观察到在MS通过的同位素峰7双峰分布。如果k CL比k 通道大得多(EX2)所观察到的速率正比至K 通道 ,由此比例常数等于该折叠-展开平衡点常数K U = K 运 </sub> / K CL。在这些条件下,许多打开和关闭事件是必要的,在对于氘核所有的酰胺质子交换,导致平均质量逐渐提高,而同位素分布保持大致相同。该EX2制度容许展开ΔGu的自由能的决心和结构元素,因此稳定性。在自然状态条件下的EX2政权是最常见的。增加了pH值和增加离液剂的可以交流机制转移到EX1。因此,HX-MS可以用来探索热力学以及蛋白质折叠和构象变化的动力学参数。
如上面提到的HX是本质的pH和温度依赖的和骨干酰胺基的溶剂完全暴露质子交换半衰期为5-400毫秒,在生理pH值(pH值7.6)和30℃,但10分钟之间,以> 15小时用在pH值2.9,平均> 2小时,0°C(除了一种多肽,它具有交换的半衰期大约为1-2分钟的第一个骨干酰胺键的质子)。在这样慢交换条件,能够使用的蛋白酶( 如胃蛋白酶),这些活性在这些条件下,与出丢失所有包含在掺入氘核的信息来消化样品。自消化道的消化缓慢交换条件下引入,全长蛋白质不仅整体HX动力学进行分析,但HX可以将其定位于特定的区域8,9。空间分辨率目前仅限于所产生的可消化性的片段,这是在一般的10-30残基之间的尺寸。然而,通过胃蛋白酶产生由于裂解的非特异性性质重叠的碎片可能会导致增加的空间分辨率。此外,其它一些蛋白酶被发现淬火条件下有活性,但是,比胃蛋白酶10少得多的效率。进一步increa空间分辨率的本身可以通过肽在气相中的分片到达由保藏的氘化图案如电子捕获解离(ECD),电子转移解离(ETD)和红外多光子解离(IRMPD)11-13的方法。这些技术避免空间分辨率的损失是由于分子内质子迁移(“加扰”),这是由碰撞诱导解离(CID)所观察到的最常用的破碎技术。然而,这些方法需要优化每个个体肽,因而仍然非常具有挑战性。
HX-MS已经被用于分析蛋白质-配体和蛋白质之间的相互作用,包括病毒衣壳组装14-17。蛋白质变性和复性,以及温度诱导的构象变化进行了调查7,18,19。磷酸化和单个氨基酸突变相关的构象变化16,20和nucleotIDE引起的变化进行了分析21,22。因此,这种方法似乎非常适合于分析组件和分子机器的动力学。一个有吸引力的候选人,其机理是非常普遍的兴趣,是Hsp90的分子伴侣复合体。
