Reductive Methylation and pH
A pH induced chemoselective reductive 13C-methylation reaction has been demonstrated on lysozyme. At low pH, the reaction prefers the N-terminal α-amine over the side chain ε-amines and vice versa at high pH. In solution the protein's amino groups exist in equilibrium between the free amine and the conjugate acid, which is tunable with pH. The optimum pH range for this reaction depends on the reducing agent used. In this case, dimethylamine borane has an optimum pH of 7.0-9.0. Working outside the optimum range of the reducing agent does make the reaction less efficient, which becomes an advantage in this case. The reductive methylation reaction preferentially modifies the N-terminal α-amine at pH 6.0 due to the lower acid dissociation constant (pKa) of the α-amine (~7)23 versus the ε-amines (~10)32. At pH 6.0 the equilibrium is drastically shifted to favor the conjugate acid; the α-amine has the highest concentration of free amine, the reactive form in the reductive methylation reaction. At pH 10.0, the equilibrium favors the free amine. Even though the α-amine with its lower pKa exists nearly entirely in the free amine form, the ε-amines are stronger bases and more reactive. In the presence of limiting 13C-formaldehyde, we are able to take advantage of the differing rates of reaction between the ε-amines and the α-amine. Reductive 13C-methylation of lysozyme at pH 10.0 with a molar ratio of 1:5 (moles of reactive amine: moles of 13C-formaldehyde) achieved methylation at each of the side chain ε-amines but not at the α-amine site, due to the weaker nucleophilic nature of the α-amine. As illustrated in Figure 2, peak 7 is absent for the lysozyme sample reacted at pH 10.0, indicating that peaks 1-6 belong to ε-dimethylamino groups and peak 7 is the N-terminal α-dimethylamino group. α-Amine favored reductive 13C-methylation at pH 6.0 was observed when the amount of 13C-formaldehyde used was reduced to a molar ratio of 2:7 and the sample was not further reacted with natural abundance formaldehyde. As illustrated in Figure 3, the methylation at low pH is a mixture of monomethylamines (13C-chemical shift ~32-34 ppm) and dimethylamines (13C-chemical shift ~43-44 ppm). Three amino groups show 13C-dimethylation, indicating that these groups reacted faster than the groups showing 13C-monomethylation. Of the 3 13C-dimethylamines, peak 7 has the highest intensity and was assigned as the N-terminal α-dimethylamino group, corroborating the assignment of peak 7 using the sample reacted at high pH.
With the ability to alter the rate of reductive methylation of the N-terminal α-amino group, the MS-assisted method for assigning the NMR resonances can be applied34. The lysozyme sample that was reductively 13C-methylated at pH 10.0 with a 5:1 ratio was digested with trypsin and analyzed with MALDI-TOF MS. Because the α-amino group was not reductively 13C-methylated under these conditions, as shown in Figure 2, the MS isotopic profile of the tetramethylated, N-terminal peptide can be used to quantify the percentage of 13C-incorporated at the N-terminal ε-dimethylamino group. The 13/12C-dimethylated N-terminal peptide, K(13/12CH3)4VFGR, with a mass-to-charge ratio (m/z) of 662, gave a scaled percentage of 13C-incorporation of 27%. NMR analysis of the same lysozyme sample gave a percentage of 13C-incorporation of 33% for peak 6, indicating that peak 6 is the N-terminal Lys1 ε-13C-dimethylamine.
Aminopeptidase and Reductive Methylation
MALDI-TOF mass spectra of untreated- and aminopeptidase-treated lysozyme samples are shown in Figure 4. Insulin, which was used as a positive control, shows a shift to lower average m/z compared to its native form, 5731.2 to 5242.7. The treated lysozyme shows a shift from 14305.8 to 13930.5, corresponding to the loss of the first three N-terminal residues of lysozyme, Lys1-Val2-Phe3. Figure 5 shows the NMR spectra of the untreated- and aminopeptidase-treated reductively 13C-methylated lysozyme samples. Compared to the control spectrum (Figure 4a), the spectrum of the aminopeptidase-treated sample (Figure 4b) shows a decrease in intensity for peaks 6 and 7 and an additional peak labeled 7a. From this data, peaks 6 and 7 can be assigned pairwise as Lys1 α- and ε-dimethylamine.
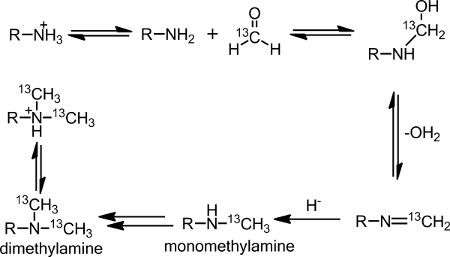
Figure 1. Reaction scheme of the reductive methylation reaction.
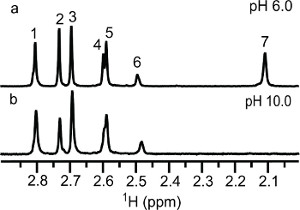
Figure 2. 1D 1H-13C HSQC NMR spectra obtained at 25 °C and pH 8.5 of reductively 13C-methylated lysozyme at reaction conditions of a 1:5 molar ratio (amine: 13C-formaldehyde) and (a) pH 6.0 and (b) pH 10.0 followed by reductive methylation with natural abundance formaldehyde in a ratio of 1:10 at pH 7.5.
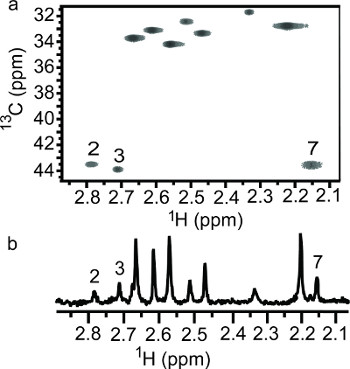
Figure 3. NMR spectra of lysozyme reductively 13C-methylated with a 2:7 molar ratio (amine: 13C-formaldehyde). (a) 2D 1H-13C HSQC and (b) 1D 1H-13C HSQC.
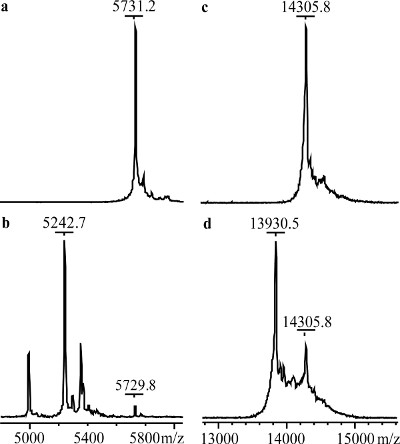
Figure 4. MALDI-TOF mass spectra. (a) Bovine pancreas insulin, (b) aminopeptidase-treated bovine pancreas insulin, (c) lysozyme, and (d) aminopeptidase-treated lysozyme.
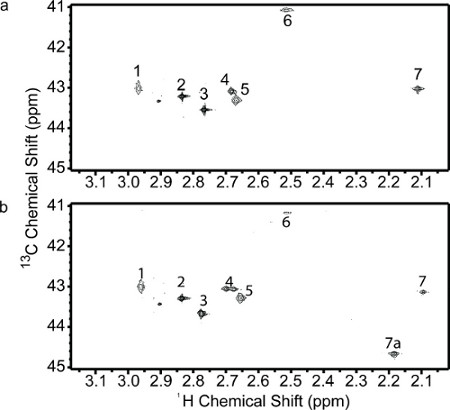
Figure 5. 2D 1H-13C HSQC NMR spectra. (a) Reductively 13C-methylated lysozyme and (b) Aminopeptidase-treated reductively 13C-methylated lysozyme.
