Figure 1 outlines the overall processing scheme for the isolation and detection of EVs using either the bead-based method or individual detection method. Individual detection of EVs using FCM works well for analyzing larger EVs but most cytometers are not capable of individually detecting particles as small as exosomes. A bead-based approach allows small EVs to be detected, however, there are drawbacks associated with using this method, as outlined in Table 1. Generally, isolation of EVs using ultracentrifugation (with or without the addition of a sucrose gradient fractionation procedure) is recommended when EVs are needed for functional assays. Ultracentrifugation removes impurities including serum proteins and other soluble contaminants from the plasma, which can affect functional experimental outcomes. However, ultracentrifugation is time consuming and may alter EV quantity and quality12.
Expected results for the two detection assays are depicted in Figures 2-3. For the individual detection assay, the lysed control (bottom row, Figure 2) is used to set gates for the corresponding non-lysed sample (top row, Figure 2). The majority of events should fall within the EV gate. Quadrant gates should not reveal double positive events when the two markers in comparison are not normally found on the same cell. The right biparameter plots in Figure 2 show the markers CD108a and CD235a, which are two red blood cell markers known to coexist on cells. Here, on EVs, over half of the positive events are positive for both markers, as expected. In the same way, cell surface markers known to reside on the same cell should show similar patterns of double positivity on EVs. The center biparameter plots show EV expression of two markers that are known not to coexist on cells. In this analysis of CD235a (a red blood cell marker) and CD41a (a platelet marker), the EVs show distinct, separate, positive populations, which is expected since they come from different cell types. When lysed, positive events should disappear. In general, any positive events remaining after lysis indicate the presence of signal coming from non-vesicle particles, aggregates, and/or detergent-resistant EVs. Figure 3 shows expected results using the bead-based detection method. Unlike the individual detection method, these data cannot/should not be viewed in bi-parameter plots. In the upper dot plots, no separation between the positive and negative populations exist, and events appear in the double positive quadrants even though they aren’t normally found on the same cell types due to the fact that both types of EVs will bind to a single bead. For the bead-based detection method, data are best analyzed using histogram overlays with the negative control (depicted underneath the dot plots). Positivity is measured using a marker’s MFI (mean fluorescence intensity) and compared directly with that of the negative control. If a sample is positive for the marker in question, its MFI will be higher than the negative control. The negative control for the bead method is simply beads blocked with BSA (no EVs added), which have been stained with the same antibodies and washed alongside the EV-coated beads. A comparison of expected results using the two methods can be seen in Figure 4.
The ability of the individual detection assay to properly assess EV phenotypes relies heavily on correct gating to separate Ab-positive events from background fluorescence. Therefore, it is critical to choose a negative control that most appropriately mimics/predicts background fluorescence for a given sample. When stained EVs are not washed before reading, commonly used negative controls (e.g., isotypes) fail to accurately predict background fluorescence for all markers (Figure 5A). In these cases, if washing is not an option, lysed samples tend to work better for predicting background fluorescence. However, when stained EVs are washed before reading (using centrifugal filtration, in this case), both negative controls (isotypes and lysed samples) work well for predicting background fluorescence of a sample (Figure 5A). It should be noted, however, that while all negative controls “work,” lysed controls are preferred because they provide additional information about a sample (e.g., the presence of detergent-resistant, non-vesicle-related events and/or aggregates) that can result in non-EV positive signals and improperly inflate Ab counts. Furthermore, isotype controls can sometimes be unreliable, even in washed samples, as shown in Figure 5B, where the stained sample has fewer positive events than the same sample stained with matched isotype control antibodies.
Without thorough removal of unbound antibody, FCM dot plots of some EV markers are nearly impossible to interpret, appearing as clouds of dimly fluorescent particles indistinguishable from their highly fluorescent backgrounds (Figure 6, top plot). Washing stained samples using centrifugal filters enhances the separation between background and positive marker signals (Figure 6, bottom plot); however, small EVs and exosomes may be lost through the pores of the filter.
The use of a detergent lysis step reveals positive, vesicle-mimicking events from immune complexes and protein aggregates21. When PPP is analyzed using individual detection, encountering positive events that do not disappear with lysis is a fairly often occurrence. These detergent-resistant events often appear as suspicious, highly fluorescent diagonal signals in both single parameter and biparameter plots (Figure 7). Clinically, these protein complexes and/or insoluble immune complexes are more prevalent in patients afflicted with various diseases21, such as rheumatoid arthritis28, nephrotic syndrome19, and systemic lupus erythematosus29. Therefore, depending on the objective of the research, one may wish to include or remove them from the analysis.Another way diagonal signals can form is by vortexing the samples, particularly after the addition of the lysis reagent (Figure 8). Samples should always be mixed up and down by pipet to prevent the formation of aggregates.
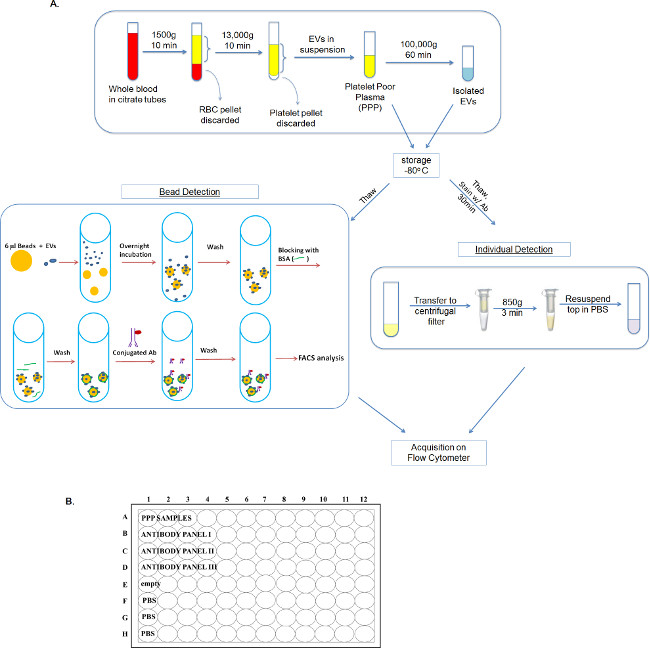
Figure 1: Overall processing scheme for the isolation and detection of EVs using either the bead-based method or individual detection method. (A) Whole blood is first processed into PPP. From there, PPP can either be processed further using ultracentrifugation to yield isolated EVs or used as-is in the individual detection or bead-based assays. (B) Outline of suggested plate map for high throughput sample analysis using the individual detection method. Please click here to view a larger version of this figure.
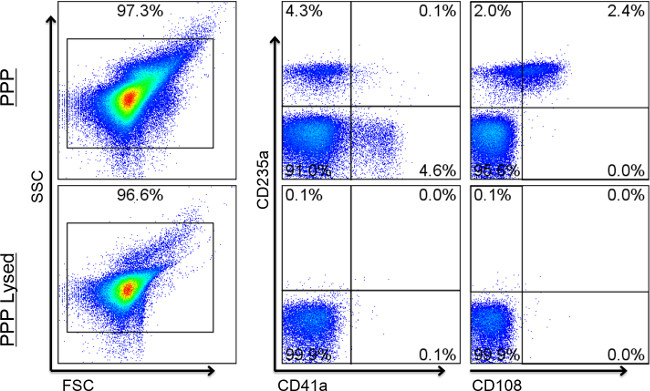
Figure 2: Expected results for Individual Detection. Flow cytometry dot plots show representative staining of lysed and unlysed EV samples. Values show percentages of positive events. The majority of events fall within the EV gate. Events shown in the right biparameter plots are within the FSC/SSC EV gates on the left. The lysed sample (bottom row) is used to set fluorescent-based gates for each corresponding (non-lysed) sample. Quad gates should not reveal double positive events when the two markers in comparison are not normally found on the same cell. Here, the biparameter CD235a and CD41a plot shows a distinct separation between the EVs expressing red blood cell markers and those expressing platelet cell markers. Likewise, cell surface markers known to reside on the same cell should show similar patterns of double positivity on EVs. The right biparameter plot shows that over half of CD235a-positive EVs are also positive for the secondary red blood cell (RBC) marker, CD108a. When lysed, positive events should disappear. Positive events remaining after lysis indicate the presence of signal coming from non-vesicle or detergent resistant particles and/or aggregates.
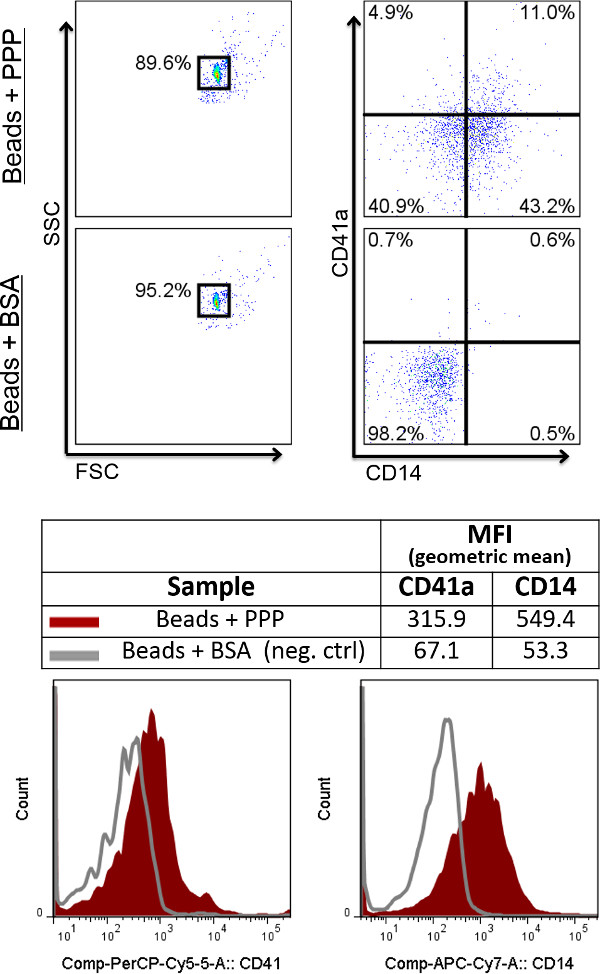
Figure 3: Expected results using the bead-based detection method. Flow cytometry dot plots show representative staining of EVs coupled to beads, as compared to beads blocked with BSA, which serves as the negative control. Values show percentages of positive events. Events shown in the right biparameter plots are within the FSC/SSC beads gates on the left. For the beads-based detection method, data are best analyzed using histogram overlays with the negative control (depicted underneath the dot plots). Positivity is measured using a marker’s MFI (mean fluorescence intensity) and compared directly with that of the negative control.
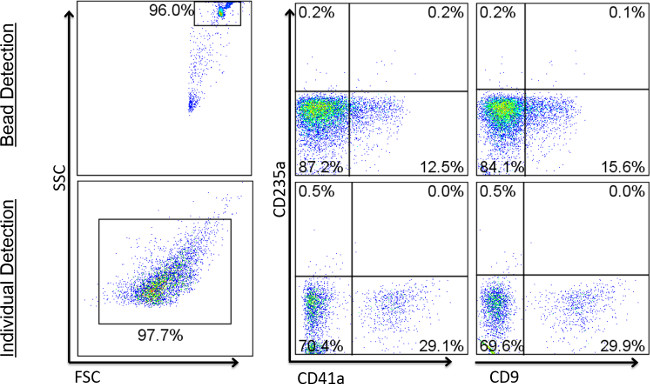
Figure 4: Comparison of expected results using beads vs. individual detection. Flow cytometry dot plots show representative staining of EVs coupled to beads (top row), compared to EVs analyzing using individual detection (bottom row). Events shown in the right biparameter plots are within the FSC/SSC beads gates on the left.
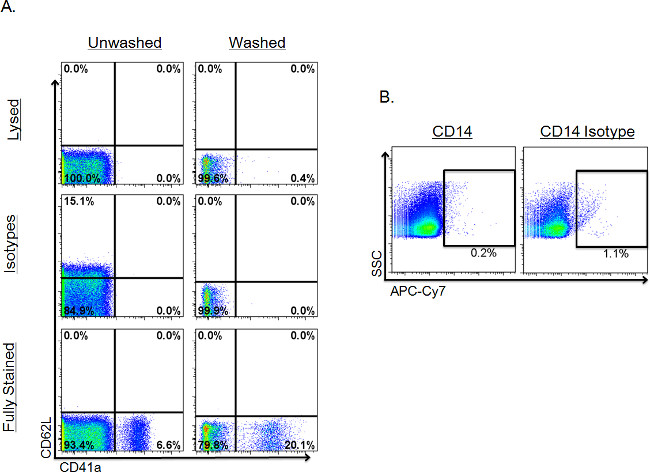
Figure 5: Comparison of different negative controls in individual detection analysis. Values show percentages of positive events. Events shown are within the FSC/SSC EV gate. (A) Comparison of negative controls in unwashed vs. washed samples. Isotype or lysed controls were evaluated for their ability to provide appropriate indications of background fluorescence across two different markers in a fully stained sample (bottom row). Gates for each marker were made using the lysed sample (top row) and then copied to the rows beneath. Washed samples were washed using post-stain filtration. (B) Example of a sample stained with the isotype control having more positive events than the sample stained with actual antibody.
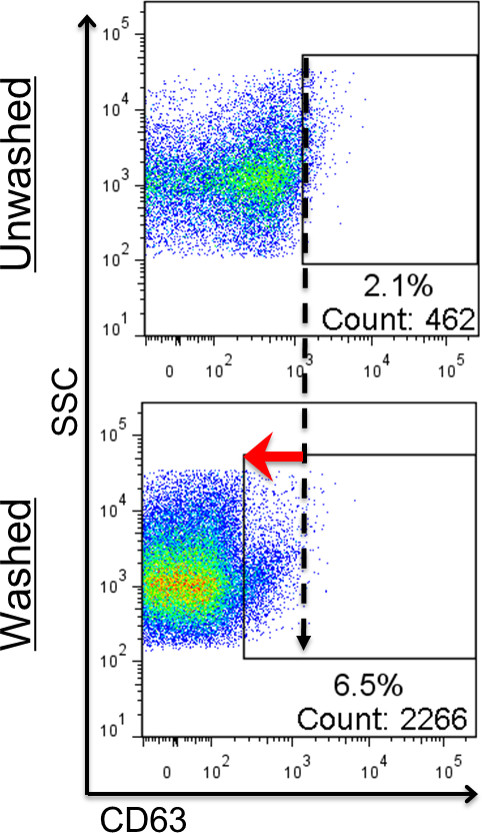
Figure 6: Effect of post-stain washing in individual detection. Values show percentages and numbers of positive events. Events shown are within the FSC/SSC EV gate. Gates for each sample were made using each sample’s lysed counterpart (not shown; refer to previous figure for gate-setting in unwashed vs washed samples). High background fluorescence makes distinguishing positive from negative events difficult (top plot). When washed, however, the positive population is revealed as unbound fluorescent antibodies are removed and background fluorescence is reduced (bottom plot).
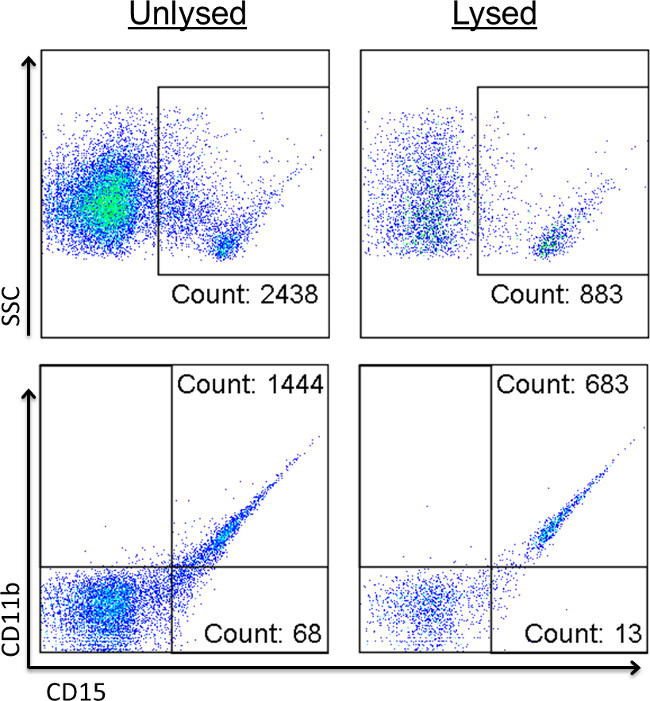
Figure 7: Detergent lysis confirms the presence of non-EV signals. Events shown are within the FSC/SCC EV gate. Values show percentages of positive events. Stained EV samples were read before (left column) and after addition of detergent (right column) to identify positive signals caused by immune complexes and other non-EV-related events.
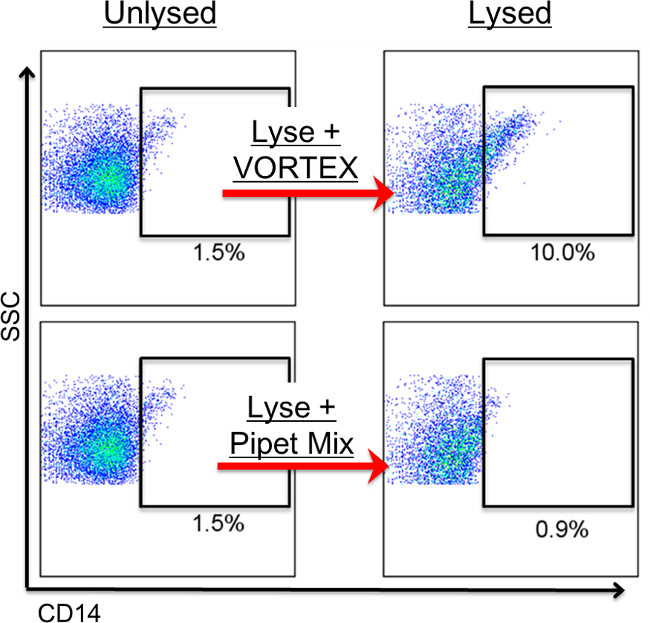
Figure 8: Vortexing causes the appearance of non-EV signals. Events shown are within the FSC/SCC EV gate. Values show percentages of positive events. Stained EV samples were read before (left column) and after addition of detergent (right column). Using a vortex to mix samples can cause EV-mimicking, diagonal populations to form (top row). When mixed gently up and down using a pipet, however, formation of these populations can be avoided (bottom row). Vortexing is not recommended for mixing, as it can cause aggregation in some samples, leading to diagonal-appearing, positive populations. In general, event number within a positive gate should not increase after lysing.
| Bead-Based Detection | Individual Detection | |
| EV Sizes | recommended for < 100 nm only | recommended for > 100 nm only |
| Time | requires overnight incubation | can complete in < 1 day |
| Sensitivity | cluster detection | single particle detection |
| Results | qualitative | quantitative |
| Washing | simple, standard centrifugation | requires centrifugal filters |
| Negative control | BSA-coated beads | lysed samples |
Table 1: Pros and cons of both detection methods.
