Analysis of Protein Import into Chloroplasts Isolated from Stressed Plants
Summary
Here we describe a new method to study protein import into isolated chloroplasts under stress. The method is rapid and straightforward, and can be applied to study the consequences of different stress conditions for chloroplast protein import, and the corresponding regulatory mechanisms.
Abstract
Chloroplasts are organelles with many vital roles in plants, which include not only photosynthesis but numerous other metabolic and signaling functions. Furthermore, chloroplasts are critical for plant responses to various abiotic stresses, such as salinity and osmotic stresses. A chloroplast may contain up to ~3,000 different proteins, some of which are encoded by its own genome. However, the majority of chloroplast proteins are encoded in the nucleus and synthesized in the cytosol, and these proteins need to be imported into the chloroplast through translocons at the chloroplast envelope membranes. Recent studies have shown that the chloroplast protein import can be actively regulated by stress. To biochemically investigate such regulation of protein import under stress conditions, we developed the method described here as a quick and straightforward procedure that can easily be achieved in any laboratory. In this method, plants are grown under normal conditions and then exposed to stress conditions in liquid culture. Plant material is collected, and chloroplasts are then released by homogenization. The crude homogenate is separated by density gradient centrifugation, enabling isolation of the intact chloroplasts. Chloroplast yield is assessed by counting, and chloroplast intactness is checked under a microscope. For the protein import assays, purified chloroplasts are incubated with 35S radiolabeled in vitro translated precursor proteins, and time-course experiments are conducted to enable comparisons of import rates between genotypes under stress conditions. We present data generated using this method which show that the rate of protein import into chloroplasts from a regulatory mutant is specifically altered under osmotic stress conditions.
Introduction
Chloroplasts are highly abundant organelles that exist in the green tissues of plants. They are well-known for their critical role in photosynthesis, a process that uses light energy to convert carbon dioxide to sugar and thus support almost all life on earth 1. In addition, chloroplasts (and the broader family of related organelles called plastids) play many other vital roles in plants, including the biosynthesis of amino acids, lipids, pigments, and the sensing of environmental signals such as gravity and pathogen challenge. Photosynthesis generates reactive oxygen species (ROS) as by-products, which under certain circumstances have useful roles, but if overproduced can cause damaging or even lethal effects. The overproduction of ROS is particularly promoted by adverse environmental conditions, and thus chloroplasts are closely linked to responses to abiotic stresses, such as salinity and osmotic stresses 2.
Chloroplasts have a complex structure. Each chloroplast is surrounded by a double-membrane outer layer called the envelope, which consists of outer and inner membranes. Internally, there is another membrane system called the thylakoids, where the light reactions of photosynthesis take place. Between the two membrane systems there is an aqueous compartment call the stroma, which is involved in carbon fixation. A chloroplast may contain up to ~3,000 different proteins, and the vast majority of these proteins are synthesized in the cytosol in precursor form and need to be imported into the organelle through dedicated protein translocons in the envelope membranes 1. Interestingly, recent work has indicated that chloroplast protein import is actively regulated, and so is able to exert an important level of control over the chloroplast proteome. For example, it was reported in 2015 that protein import can respond to abiotic stress through the direct regulation of the abundance of the translocon at the outer envelope membrane of chloroplasts (TOC) by the ubiquitin-proteasome system 3.
Using purified chloroplasts and in vitro synthesized precursor proteins, protein import can be reconstituted in vitro 4,5. Thus, in vitro methods can be used to assess the rates of import in different mutant plants 6, which has been a critical approach for the analysis of putative components of the protein import machinery and for discovering the mechanisms underlying protein import and its regulation. Moreover, chloroplasts can be processed with further fractionation or protease digestion, following in vitro import, which can facilitate studies on the sub-organellar localization and topology of chloroplast proteins 7,8.
To study the regulation of protein import by stress, we have modified our routine chloroplast isolation method as we will describe here. Importantly, chloroplasts were isolated from plants that had been grown on standard Murashige and Skoog (MS) agar medium for 8 days and then transferred into liquid MS medium supplemented with stressor, providing a relatively short, controlled stress treatment. The yield and competency of chloroplasts isolated from such stress-treated plants are compatible with the downstream in vitro protein import assay 3. In addition to the chloroplast isolation protocol, we present our routine method for in vitro protein import, which has proven to be robust and is widely used 3,9-12.
Protocol
1. Growth of Arabidopsis Plants and the Stress Treatment
- Prepare 1 L of Murashige and Skoog (MS) medium by adding 4.3 g of MS Basal Salt Mixture, 10 g sucrose, 0.5 g 2-(N-morpholino) ethane sulfonic acid (MES) to deionized water up to 1 L, and adjust the pH to 5.7 with potassium hydroxide (KOH). Add 6 g of phytoagar and autoclave for 20 min at 120 °C.
- Before solidification, pour the medium into round Petri plates (diameter 9 cm, height 1.5 cm, with 20 – 25 mL MS medium per plate). Allow the plates to dry for ~1 h in a laminar flow hood before closing the lids.
- Place Arabidopsis thaliana seeds into a 1.5 mL test tube for surface sterilization by adding 1 mL of 70% (v/v) ethanol containing 0.02% (v/v) Triton X-100 and continuously shaking for 5-10 min. For each genotype/condition, prepare 10 Petri plates each carrying ~100 – 150 seedlings.
- Wait for about 10 s until the seeds settle to the bottom of the tube, and then discard the supernatant by pipetting. Add 1 mL of 100% ethanol, and shake the tube again for 10 min.
- Prepare the laminar flow hood while the seeds are sterilizing. Take one piece of filter paper per sample, fold it in half to facilitate sowing, and soak it in 100% ethanol in the hood. Wait until it dries out completely. Note that 100% ethanol is used as it evaporates more quickly than 70% ethanol.
- Transfer the seeds onto the filter paper by pipetting using a 1 mL pipette tip cut ~5 mm from the fine end. Allow them to dry, which should take about 10 – 15 min.
- Sow ~100 – 150 sterilized seeds evenly onto each MS medium Petri plate, and seal each plate with surgical tape.
- Store the plates upside down at 4 °C for 2 – 4 d to encourage and synchronize germination. Old seeds may need to stay longer (up to a week) at 4 °C.
- Transfer the plates to a plant tissue culture chamber and leave them upside up. Grow the plants for 8 d under a long-day cycle (16 h 100 µmol·m−2·s−1 light, 8 h dark) at 20 °C.
- For stress treatment, when the plants are 8 d old, in the flow hood, transfer them from the agar medium, by gently scraping them off by hand wearing ethanol-sterilized gloves, into a sterilized flask of liquid MS medium containing the stressor (e.g., 200 mM mannitol). Avoid carrying over agar medium. Cover the flask mouth with sterilized foil and allow the plants to grow under the same conditions as in step 1.8 for an additional 2 d on an orbital shaker with gentle shaking (~100 rpm).
2. Making a Precursor Protein by In Vitro Transcription/Translation
Note: This protocol assumes the use of Arabidopsis photosystem I subunit D precursor (pPsaD) as the template/preprotein, but the method is compatible with others.
- Clone the coding sequence (CDS) of pPsaD into the multiple cloning site (MCS) of the pBlueScript II SK vector (or any other similar vector with an upstream T7 promoter) 13. Purify the plasmid (pBSK-pPsaD) using a DNA isolation kit and verify the sequence by DNA sequencing.
NOTE: All these steps employ standard molecular cloning techniques. - Determine the pBSK-pPsaD plasmid DNA concentration using a spectrophotometer set to measure absorbance at 260 nm. Dilute the plasmid to a final concentration of 10 ng/µL.
- Run a 20 µL PCR (for 35 cycles) using the diluted plasmid as template. Prepare the reaction as follows: 2 µL 10× polymerase buffer, 2 µL 2 mM dNTPs, 1 µL 5 mM M13 forward primer (5'-TGT AAA ACG ACG GCC AGT-3'), 1 µL 5 mM M13 reverse primer (5'-CAG GAA ACA GCT ATG ACC-3'), 1 µL diluted pBSK-pPsaD plasmid, 1 U Taq polymerase, and distilled water to make the total reaction volume up to 20 µL. Use a standard PCR program, as follows: 95 °C, 5 min; 35 cycles of [95 °C, 30 sec; 56 °C, 30 sec; 72 °C, 30 sec); and 72 °C, 5 min.
- Run 5 µL of the PCR product on a 1% (w/v) agarose gel in TAE buffer (Tris-HCl, pH 7.6, 20 mM acetic acid, 1 mM EDTA) to verify correct amplification of the cDNA, and quantify the relevant band relative to standards to confirm the concentration. Store the rest of the product at −20 °C for further applications.
- Prepare a 50 µL reaction using a rabbit reticulocyte lysate based cell-free transcription/translation system compatible with PCR DNA, as follows: 40 µL reticulocyte lysate from the transcription/translation system, 2.5 µL radiolabeled [35S] methionine, 11 µCi/mL (specific activity: >1,000 Ci/mmol), 2.5 µL sterile distilled water, and 5 µL pPsaD PCR product (100 – 800 ng).
Caution: Wear gloves, laboratory clothing and safety glasses when handling radioactive material. Monitor and decontaminate the working surface and equipment. Dispose of all radioactive waste in an approved waste container. - Incubate the reaction for 90 min in a 30 °C water bath. Stop the reaction by placing the sample on ice.
- Remove 1 µL of the reaction as a test sample (the same volume can also serve as an input control for step 5.7) for verification on standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) followed by autoradiography, fluorography or phosphor imaging. A good result is indicated by the observation of a distinct and strong band corresponding to the molecular weight of pPsaD (~23 kD). Store the rest of the reaction at −80 °C for further applications.
3. Chloroplast Isolation
- Prepare the following stock solutions:
- Prepare Chloroplast Isolation Buffer (CIB, 2x): 0.6 M sorbitol, 10 mM magnesium chloride (MgCl2), 10 mM Ethylene Glycol Tetraacetic Acid (EGTA), 10 mM ethylenediaminetetraacetic acid (EDTA), 20 mM sodium bicarbonate (NaHCO3), 40 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES); adjust to pH 8.0 with KOH. Prepare 2 L of 2x CIB, make aliquots and keep them at -20 °C for long-term storage, or at 4 °C for short-term storage. To make 1x CIB, dilute the 2x CIB 1:1 with distilled water; this can be prepared freshly on the day of chloroplast isolation experiment.
- Prepare HEPES-MgSO4-Sorbitol buffer (HMS, 1x): 50 mM HEPES, 3 mM magnesium sulfate (MgSO4), 0.3 M sorbitol; adjust to pH 8.0 with sodium hydroxide (NaOH). Prepare 400 mL of buffer, make aliquots and keep them at -20 °C for long-term storage, or at 4 °C for short-term storage.
- Conduct all of the following procedures in the cold room or on ice. Precool all of the solutions, devices, equipment, and rotors before starting.
- Prepare a continuous density gradient by mixing 13 mL Percoll, 13 mL 2x CIB buffer, and 5 mg glutathione in a 50 mL centrifuge tube, and centrifuging the mixture at 43,000 × g for 30 min (brake off) at 4 °C. After centrifugation, handle the tube with care to avoid disturbing the gradient and keep it on ice for later use.
- Transfer 100 mL of 1x CIB per genotype/condition to a 1 L beaker.
- Remove the seedlings from the liquid medium by tipping them into a sieve, and transfer them into another CIB-containing beaker; then, rinse the tissue with the CIB to remove any residual liquid medium before replacing it with 100 mL of fresh CIB.
- For homogenization, use a total of 100 mL of 1x CIB per sample in five consecutive rounds of homogenization, each round using 20 mL of fresh CIB held in a 50 mL beaker.
- For the first round of homogenization, transfer the plant tissue into the 50 mL beaker by hand, allowing the previous holding buffer to drain through your fingers. Try to use most of the tissue in the first round, but make sure it is immersed with buffer; if this is not possible, introduce the remaining unused tissue at the second round.
- Place probe of the tissue homogenizer into the tissue and homogenize with two pulses of 1 – 2 s each. Filter the homogenate through two layers of filtration cloth into a 250 mL centrifuge tube by gentle squeezing. Retain the filtrate, and transfer the tissue back to the 50 mL beaker.
- Place a second 20 mL CIB aliquot into the 50 mL beaker, adding any remaining tissue not used in the first round of homogenization, and repeat the homogenization and filtration steps. Thus, repeat steps 3.5 and 3.6 until all of the 100 mL of CIB has been used up (i.e., 5 aliquots of 20 mL), and combine the resulting filtrates.
- Centrifuge the pooled homogenate at 1,000 × g for 5 min (brake on) at 4 °C, and pour off the supernatant immediately following completion of the centrifugation, taking care not to disturb the pellet. Resuspend the pellet in the residual supernatant left in the tube by gently agitating the tube on ice. Do not resuspend vigorously by pipetting or vortexing.
- Gently transfer the homogenate onto the top of the premade continuous density gradient, using a Pasteur pipette to apply it via the wall of the receiving tube. Avoid disturbing the gradient. Centrifuge in a swinging-bucket rotor at 7,800 × g for 10 min (brake off) at 4 °C.
NOTE: Two green bands can be seen in the gradient after centrifugation: the lower band contains intact chloroplasts, and the upper band contains broken chloroplasts. - Discard the top band by pipetting, and then transfer the lower band with a Pasteur pipette into a fresh 50 mL centrifuge tube, retaining a volume of up to 8 mL per gradient.
- Add ~25 mL of 1x HMS buffer into the tube and invert the tube twice to wash the Percoll from the chloroplasts. Place the tube again in the swinging-bucket rotor and centrifuge at 1,000 × g for 5 min (brake on) at 4 °C.
- Pour off the supernatant and carefully resuspend the chloroplast pellet in the residual HMS left in the tube by gently agitating the tube on ice. Add an additional 100 – 300 µL of HMS if necessary (based on the size of the pellet and the counting in Section 4), but try not to dilute the sample too much.
- Keep the chloroplasts on ice. Each time before the chloroplasts are used for downstream applications, shake the tube to resuspend them. Always use a cut pipette tip (with an enlarged pore) to transfer the chloroplasts.
4. Analysis of the Yield and Intactness of Chloroplasts
- Add 5 µL of isolated chloroplasts to 495 µL of 1x HMS buffer in a 1.5 mL tube, and then mix gently by inverting the tube to obtain a 1:100 dilution.
- Place a cover glass on top of the counting chamber of the haemocytometer. Slowly pipette ~40 – 60 µL of the diluted chloroplast suspension into the gap between the cover glass and the counting chamber. Using a phase-contrast microscope with a 10X or 20X objective, intact chloroplasts look round and bright and are surrounded by a halo of light.
NOTE: Under the microscope, there is a 1 mm2 counting area with 25 large squares, each containing 16 small squares in the center of the counting chamber. - Count the number of chloroplasts in 10 large squares. The number of chloroplasts per large square should average between 10 and 30. If too few or too many chloroplasts are present, adjust the dilution factor (step 4.1 above) accordingly, and repeat the procedure.
- Calculate the number of chloroplasts per mL (concentration) as follows: n (the average number of chloroplasts per large square calculated in step 4.3) × 25 (total number of large squares) × 100 (the dilution factor) × 104 (scaling factor to express data per 1 mL, since the volume above the 25 squares is 0.1 mm3).
- Calculate the actual yield of chloroplasts by multiplying the concentration by the volume of chloroplast suspension obtained in step 3.12. Normally, more than 50 × 106 chloroplasts can be obtained.
5. Chloroplast Protein Import
- Prepare the following stock solutions:
- Prepare 10x HMS buffer: 500 mM HEPES, 30 mM MgSO4, and 3.0 M sorbitol; adjust to pH 8.0 with NaOH. Prepare 50 mL of this buffer, aliquot, and store at −20 °C for long-term storage and at 4 °C for short-term storage.
- Prepare Import stop buffer: 50 mM EDTA dissolved in 1x HMS buffer. Prepare 50 mL of this buffer, aliquot, and store at −20 °C.
- Prepare 2x Protein loading buffer: 60 mM Tris-HCl, pH 6.8, 10% (v/v) glycerol, 2% (w/v) SDS, 0.005% (w/v) bromophenol blue. Make 50 mL, and store at 4 °C. Just before use, add 100 µL of 1 M 1,4-dithiothreitol (DTT) to 900 µL of buffer.
- To run a time-course with 3 time-points, prepare 3 tubes each containing a 130 µL aliquot of import stop buffer, and leave them on ice. Prepare a 450 µL import reaction in a 2 mL tube (per genotype/condition). Thaw all ingredients just prior to use.
- For one import reaction, typically use 10 × 106 chloroplasts in A µL volume; for example, if the chloroplast concentration is 2.5 × 108 /mL, use 40 µL chloroplast suspension. Three time-points will need 3 × A µL (i.e., 120 µL in our example).
- Mix the reactions components on ice in the following order: distilled water (to make the total volume 600 µL), B µL 10x HMS buffer (where B = [600-3×A]/10; i.e., 48 in our example), 12 µL 1 M gluconic acid (potassium salt), 6 µL 1 M NaHCO3, 6 µL 20% (w/v) BSA, 30 µL 100 mM adenosine 5′-triphosphate magnesium salt (MgATP), 24 µL 250 mM methionine (not radiolabeled), and 30 µL precursor protein. Immediately before starting the import reaction, add 3 × A µL of chloroplasts and mix by gently tapping the tube.
- Incubate the reaction tube at 25 °C in a water bath under 100 µmol·m−2·s−1 light. Occasionally flick the tubes to resuspend chloroplasts.
- To conduct a time-course, withdraw 130 µL aliquots from the reaction at the required time-points within the linear range of import, which for pPsaD is up to ~12 min (4-, 8- and 12-min time points are suitable in this case). The linear range period can vary for different proteins 14. Thus, it is suggested to test each individual preprotein before to optimize the time points.
- Immediately upon withdrawal, transfer each 130 µL aliquot to a tube of ice-cold import stop buffer, mix by gently tapping the tube, and retain all tubes on ice until the time-course has been completed.
- Centrifuge all samples for 30 s at 12,000 × g in a microfuge, discard the supernatants by pipetting, and resuspend pellets in 15 µL of 2x Protein loading buffer by vortexing.
- Analyze all the samples plus the pPsaD input control (containing pPsaD equivalent to 10% of the amount added to each import reaction, from step 2.7) by standard SDS-PAGE and autoradiography, fluorography or phosphor imaging 15. Use image analysis software to quantify and analyze the results. To provide an indication of import efficiency, the amount of imported protein in different genotypes/conditions can be assessed by measuring the radioactivity associated with each mature band.
Representative Results
An example chloroplast protein import experiment with 3 time-points is shown in Figure 1. PsaD is an ~18 kD component of photosystem I exposed to the stroma, with a precursor form of ~23 kD16. PsaD was selected here for the in vitro protein import assay because its steady-state levels are elevated in the sp1 mutant, relative to WT, under stress conditions, suggesting a change in its import efficiency in the mutant 3. The sp1 mutant carries a defect in an important regulator of the chloroplast protein import machinery — the SP1 protein 3. For chloroplasts isolated from plants grown under normal conditions, there was no obvious difference in PsaD import between sp1 and WT (data not shown)3. However, using the methods described here to assess PsaD import into chloroplasts isolated from osmotically stressed plants, a clear difference was detected. While we observed the accumulation of the mature protein form in a time-dependent manner with both genotypes, the rate of import was significantly lower for WT chloroplasts than for sp1 chloroplasts (Figure 1), which is consistent with our previous results 3, and reveals an important role for the SP1 protein in regulating the chloroplast import of PsaD under stress conditions.
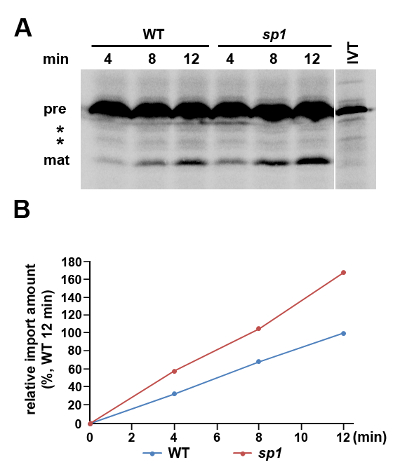
Figure 1. A Protein Import Assay Conducted using Chloroplasts Isolated from Plants Grown under Osmotic Stress Conditions. Chloroplasts were isolated from a 10-day-old WT (Col-0) and a sp1 mutant Arabidopsis plants grown under stress conditions (200 mM mannitol) for 2 d. (A) Protein import was conducted using [35S]-methionine-labeled pPsaD and allowed to proceed for 4, 8 and 12 min before analysis by SDS-PAGE and phosphor imaging. In parallel, the pPsaD 10% input control comprising in vitro translated protein (IVT) was analyzed. The precursor (pre) and mature (mat) forms of pPsaD are indicated, while in between there are two bands that likely correspond to truncated or proteolyzed translation products because their intensity did not change during the time course (*). (B) To compare the rates of import into chloroplasts from WT and sp1 plants grown under stress conditions, the intensity of each band corresponding to imported mature protein in A was quantified. All data are expressed as percentages of the amount of imported protein in chloroplasts from WT plants after 12 min. Please click here to view a larger version of this figure.
Discussion
We recently showed that chloroplast protein import can be actively regulated by stress, which is critical for chloroplast function and plant survival 3. In that study, to monitor such regulation, we modified our chloroplast isolation and in vitro import assay procedures in order to enable assessment of the import capacity of plants grown under stress conditions. The results indicated an important role for SP1 in chloroplast protein import regulation.
Conventional in vitro import assays use plants grown on standard MS agar medium6,17,18. In the case of the sp1 mutant described here, such conventional assays did not reveal any differences in protein import relative to the WT3. However, the role of SP1 in regulating protein import is clearly revealed when protein import is assessed under stress conditions using the methods described herein (Figure 1). While it may not be possible to directly compare import data from the stress conditions assay with those obtained from conventional assays (as the methods employ plants grown in liquid culture and on agar medium, respectively), comparisons between stress and non-stress conditions are feasible provided that the plants are all grown in the same liquid culture medium, with or without stressors.
Compared with the stress treatment on agar medium, liquid culture is more convenient for the treatment of the large numbers of plants needed for in vitro import assays. Moreover, it facilitates the uniform application of the stressor to all plants, which is particularly important for short-term stress treatments. The method presented here has been applied to study the osmotic stress using mannitol treatment, but could easily be adapted to a wide range of other stresses; for example, short-term salt stress and oxidative stress, for which the corresponding stressors could be similarly applied via liquid MS medium. For other types of stresses, we suggest the degree of stress is first optimized; overly severe treatments may have adverse effects on the yield and/or import competence of the isolated organelles.
There are several important steps within the protocol to which one should pay special attention, as detailed below.
The optimal agar concentration for the MS medium can differ (0.6 – 0.9%, w/v) depending on the manufacturer. Thus, it is recommended to empirically optimize the agar concentration before commencing experiments. The medium should not be so soft that it sticks to the tissue at the harvesting step (step 1.9), neither should it be so hard that it inhibits plant root development. The sucrose concentration can also be adjusted according to the plants used. When working with particularly sick mutants, MS medium supplemented with 2 – 3% (w/v) sucrose may help plants to grow better. When applying stress treatments, it is not good to transfer very old plants to the liquid medium (e.g., >14 days old). This is because the more developed roots of older plants are more easily damaged during transferral.
With regard to the transcription/translation system, there are 2 major systems: those based on wheat germ, and those based on rabbit reticulocyte. These kits may use different templates, such as linearized plasmids, unlinearized plasmids, or PCR products. The kits are also specific for the T3, T7, and SP6 promoters. Note that the kit we recommend here is only suitable for use with PCR products and the T7 promoter. However, for unknown reasons, some radiolabeled preproteins made with this system might not work efficiently in the import assay; in such instances, one may consider trying a wheat germ extract system or a reticulocyte lysate system meant for plasmid templates. One may also improve the result of the transcription/translation reaction by modifying the reaction conditions according to the manufacturer's handbook.
It is important to start chloroplast isolation early in the morning (or early in the light cycle of the growth chamber) in order to avoid accumulation of starch inside the chloroplasts due to photosynthesis, which can hinder the isolation of intact organelles. The chloroplast isolation procedure has to be done quickly without any unnecessary delays, and the isolated chloroplasts must always be kept cold. This is to mitigate against the observation that isolated chloroplasts will gradually lose their viability, which is not good. If using newly thawed CIB or HMS buffer, be sure to mix the buffer well before use to obtain a homogeneous solution. The optimal conditions for the homogenization of plant material have been established empirically, and can vary if a different tissue homogenizer is used.
If the different plant genotypes contain similar chlorophyll levels, chlorophyll quantification can be used as an alternative way to normalize the samples before conducting the import assays. Chlorophyll can be determined spectrophotometrically following extraction of a sample of the isolated chloroplasts in 80% (v/v) aqueous acetone 19,20. However, if the import rates of plants showing different chlorophyll contents (e.g., mutants with chlorotic phenotypes) are to be compared, use chloroplast number counting to normalize the chloroplast samples in the import assays. It is particularly important to resuspend the chloroplasts thoroughly in step 3.11. Insufficient resuspension may leave aggregates of chloroplasts, which will make it difficult to count numbers accurately and thus will hinder the correct loading in import reactions. If severe aggregation is seen under the microscope (e.g., aggregates with >10 chloroplast joined together), continue shaking the chloroplast sample on ice until the aggregates are removed. It is easier to resuspend the chloroplasts in a smaller volume of buffer.
It is important to be aware that protein import reactions (Section 5) must be conducted with appropriate precautions because of their radioactive nature. Necessary precautions include: wearing disposable gloves, laboratory clothing and safety glasses, monitoring and decontaminating the working surface and equipment, and disposing of all radioactive waste in an approved waste container. Also keep in mind that the correct osmotic pressure is critical for maintaining the intactness of chloroplasts during the import reactions, and this is chiefly maintained by the HMS buffer. Because 10x HMS is viscous, it should first be warmed up to RT, and then thoroughly mixed, and applied using a cut pipette tip to ensure measurement of accurate volumes. In the import reaction, cold methionine is added to inhibit the incorporation of free radiolabeled methionine into unrelated chloroplast proteins through organellar translation during the incubation step, while BSA is used to minimize proteolysis by acting as a substrate for proteases.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by a grant to PJ from the Biotechnology and Biological Sciences Research Council (BBSRC; grant ref. BB/K018442/1).
Materials
| Murashige and Skoog basal salt | Melford | M0221 | |
| phytoagar | Melford | P1003 | |
| 2-(N-morpholino) ethane sulfonic acid (MES) | Melford | B2002 | |
| triton X-100 | Fisher | BPE151-500 | |
| surgical tape (e.g., Micropore, 3M) | 3M | 1530-1 | |
| filter paper | Fisher | FB59023 | |
| Percoll | Fisher | 10607095 | |
| ethylene glycol tetraacetic acid (EGTA) | Sigma | E4378 | |
| ethylenediaminetetraacetic acid (EDTA) | Fisher | D/0700/53 | |
| 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) | Melford | B2001 | |
| filtration cloth (Miracloth) | Calbiochem | 475855 | |
| 50 mL centrifuge tube | Fisher | CFT-595-040M | |
| 250 mL centrifuge bottle | Fisher | CFT-891-V | |
| Polytron (e.g., Kinematica PT10-35) | Fisher | 11010070 | |
| Polytron probe (e.g., Kinematica PTA20S) | Fisher | 11030083 | |
| centrifuge (e.g., Beckman Coulter Avanti JXN-26, for 50 and 250 mL tubes) | Beckman Coulter | B34182 | |
| fixed-angle rotor for 250 mL bottles (e.g., JLA-16.250) | Beckman Coulter | 363930 | |
| fixed-angle rotor for 50 mL tubes (e.g., JA-25.50) | Beckman Coulter | 363058 | |
| swinging-bucket rotor for 50 mL tubes (e.g., JS-13.1) | Beckman Coulter | 346963 | |
| radiolabeled [35S] methionine | Perkin Elmer | NEG072002MC | |
| rabbit reticulocyte lysate based cell-free translation system (TNT T7 Quick kit for PCR DNA) | Promega | L5540 | |
| phase-contrast microscope (e.g., Nikon Eclipse 80i) | Nikon | unavailable | |
| haemocytometer (Improved Neubauer BS748 chamber) | Hawksley Technology | AC1000 | 0.1 mm depth, 1/400 mm2 |
| cover glasses | VWR | 16004-094 | 22 mm × 22 mm, thickness 0.13-0.17 mm |
| 1.5 mL microfuge tubes | Sarstedt | 72.690.001 | |
| 2 mL microfuge tubes | Starlab | S1620-2700 | |
| microfuge (e.g., Eppendorf 5415D) | Eppendorf | unavailable | |
| Nanodrop 2000 spectrophotometer or similar | Thermo Fisher | SPR-700-310L | |
| gluconic acid (potassium salt) | Fisher | 22932-2500 | |
| bovine serum albumin (BSA) | Sigma | A7906 | |
| MgATP | Sigma | A9187 | |
| methionine | Sigma | M6039 | |
| bromophenol blue | Fisher | B/P620/44 | |
| glycerol | Fisher | G/0650/17 | |
| SDS | Fisher | S/5200/53 | |
| Tris Base | Melford | B2005 | |
| dithiothreitol (DTT) | Melford | MB1015 | |
| image analysis software (e.g., Aida Image Analyzer) | Raytest | unavailable |
References
- Jarvis, P., Lòpez-Juez, E. Biogenesis and homeostasis of chloroplasts and other plastids. Nat. Rev. Mol. Cell Biol. 14 (12), 787-802 (2013).
- Saibo, N. J., Lourenco, T., Oliveira, M. M. Transcription factors and regulation of photosynthetic and related metabolism under environmental stresses. Ann. Bot. 103 (4), 609-623 (2009).
- Ling, Q., Jarvis, P. Regulation of chloroplast protein import by the ubiquitin E3 ligase SP1 is important for stress tolerance in plants. Curr Biol. 25 (19), 2527-2534 (2015).
- Chua, N. H., Schmidt, G. W. In vitro synthesis, transport, and assembly of ribulose 1,5-bisphosphate carboxylase subunits. Basic Life Sci. 11, 325-347 (1978).
- Highfield, P. E., Ellis, R. J. Synthesis and transport of the small subunit of chloroplast ribulose bisphosphate carboxylase. Nature. 271, 420-424 (1978).
- Aronsson, H., Jarvis, P. A simple method for isolating import-competent Arabidopsis chloroplasts. FEBS Lett. 529 (2-3), 215-220 (2002).
- Froehlich, J. Studying Arabidopsis envelope protein localization and topology using thermolysin and trypsin proteases. Methods Mol Biol. 774, 351-367 (2011).
- Flores-Pérez, &. #. 2. 1. 8. ;., Jarvis, P. Isolation and suborganellar fractionation of Arabidopsis chloroplasts. Methods Mol. Biol. 1511, 45-60 (2017).
- Kubis, S., et al. The Arabidopsis ppi1 mutant is specifically defective in the expression chloroplast import, and accumulation of photosynthetic proteins. Plant Cell. 15 (8), 1859-1871 (2003).
- Kubis, S., et al. Functional specialization amongst the Arabidopsis Toc159 family of chloroplast protein import receptors. Plant Cell. 16 (8), 2059-2077 (2003).
- Aronsson, H., et al. Nucleotide binding and dimerization at the chloroplast pre-protein import receptor, atToc33, are not essential in vivo but do increase import efficiency. Plant J. 63 (2), 297-311 (2010).
- Huang, W., Ling, Q., Bédard, J., Lilley, K., Jarvis, P. In vivo analyses of the roles of essential Omp85-related proteins in the chloroplast outer envelope membrane. Plant Physiol. 157 (1), 147-159 (2011).
- Sambrook, J., Fritsch, E. F., Maniatis, T. . Molecular Cloning: A Laboratory Manual. Second edn. 1, (1989).
- Aronsson, H., et al. Monogalactosyldiacylglycerol deficiency in Arabidopsis thaliana affects pigment composition in the prolamellar body and impairs thylakoid membrane energization and photoprotection in leaves. Plant Physiol. 148, 580-592 (2008).
- Aronsson, H., Jarvis, R. P. Rapid isolation of Arabidopsis chloroplasts and their use for in vitro protein import assays. Methods Mol. Biol. 774, 281-305 (2011).
- Haldrup, A., Lunde, C., Scheller, H. V. Arabidopsis thaliana plants lacking the PSI-D subunit of photosystem I suffer severe photoinhibition, have unstable photosystem I complexes, and altered redox homeostasis in the chloroplast stroma. J. Biol. Chem. 278 (35), 33276-33283 (2003).
- Kubis, S. E., Lilley, K. S., Jarvis, P. Isolation and preparation of chloroplasts from Arabidopsis thaliana plants. Methods Mol. Biol. 425, 171-186 (2008).
- Chen, X., Smith, M. D., Fitzpatrick, L., Schnell, D. J. In vivo analysis of the role of atTic20 in protein import into chloroplasts. Plant Cell. 14, 641-654 (2002).
- Miras, S., et al. Non-canonical transit peptide for import into the chloroplast. J. Biol. Chem. 277 (49), 47770-47778 (2002).
- Nada, A., Soll, J. Inner envelope protein 32 is imported into chloroplasts by a novel pathway. J. Cell Sci. 117 (17), 3975-3982 (2004).
