Overview of In Vitro Differentiation of hPSCs
This in vitro differentiation protocol begins with undifferentiated hESCs grown on MEFs that are transitioned to feeder-free conditions for one passage (Figure 1A). While we described the differentiation of hESCs in this protocol, we used this protocol to successfully differentiate hiPSCs into trophoblastic cells. The transition to extracellular matrix removes the majority of the irradiated MEFs, which are undesirable for analyses that require pure populations of human trophoblastic cells. Undifferentiated hPSCs are grown on extracellular matrix and cultured with CM containing B-FGF until the colonies are ready for passaging (approximately one week). This maintains the pluripotent state prior to inducing BMP4/A/P-mediated differentiation. The colonies are disrupted into clumps of ~50-100 cells using dispase and passaged a second time onto an extracellular matrix-coated plate. The day after passaging, the adherent cells are ready to induce differentiation, and the medium is switched to contain BMP4/A/P lacking B-FGF. The differentiated cells proliferate for approximately 2 weeks, during which time the cells can be collected for RNA isolation or used for transfection experiments. Morphological changes appear 1-2 days after differentiation has been initiated, and results from one representative experiment are shown in Figure 1B. Notice that cells on differentiation days 1-2 are larger in size than the cells from day 0 (Figure 1B). The differentiated cell colony grows rapidly, and this change is noticeable every day. As differentiation proceeds, the cells divide and expand out from the center of the colony, growing in an outward direction (days 3-5; Figure 1B). The outer portion of the colony contains larger cells compared to the cells at the center of the colony. The differentiating cells become darker and flatter than pluripotent stem cells cultured on extracellular matrix; the changes in cell brightness are more apparent when viewing the cells with an upright microscope used for routine cell culture. The individual colonies merge together by differentiation day 5 (Figure 1B), with cells growing on top of each other. After differentiation day 4-5, cells containing multiple nuclei are present (not shown) (Figure 1B).
BMP4/A/P Induces the Differentiation and Expression of Trophoblast Markers
We examined gene expression changes during the first three days of BMP4/A/P differentiation using quantitative RT-PCR (qRT-PCR). RNA was isolated at differentiation days 1, 2, and 3 and converted to cDNA. The disappearance of pluripotent stem cells can be assessed both by morphology, visually using a microscope, and by qRT-PCR, using primers for pluripotency genes. The relative expression levels of NANOG, a marker of pluripotent stem cells, is reduced by ~75% after one day of differentiation when cells are cultured with BMP4, and levels are reduced by ~90% in the presence of BMP4/A/P (Figure 2). By differentiation day 2, the levels of NANOG are very low for cells cultured in BMP4 alone or in BMP4/A/P compared to those in hESC CM medium (containing B-FGF). This is an expected result, because we do not observe undifferentiated cells after two days of differentiation (Figure 1B), which are brighter and smaller in size compared to differentiated cells. The efficiency of BMP4-mediated differentiation to trophoblastic cells can be directly assessed by qRT-PCR using primers specific for two trophoblast markers: KRT7 and CDX2. Both of these genes are not expressed in human pluripotent stem cells, and their expression increases after 2-3 days of BMP4 treatment (Figure 2). Using the conditions described here, KRT7 transcript levels increase on differentiation day 1 in BMP4/A/P medium and remain constant for the first 3 days of differentiation (Figure 2). KRT7 expression when using BMP4 alone has a delayed increase by day 2, in agreement with previous observations that Activin/Nodal inhibitors increase the differentiation rate of hESCs9. Another way to assess the efficiency of using these inhibitors is to determine the steady-state abundance of CDX2 and KRT7 transcripts in cells treated with BMP4 alone. CDX2 levels are similar for differentiation days 1-3 when using either BMP4 alone or BMP4 together with Activin/Nodal inhibitors. However, KRT7 transcripts are more abundant after one day of differentiation in the presence of these inhibitors, which suggests a more rapid differentiation (Figure 2). CDX2 expression typically peaks at differentiation day 2 for both conditions and decreases after day 3 (Figure 2). Undifferentiated hESCs do not express either KRT7 or CDX2 (Figure 2), as expected. In conclusion, BMP4/A/P conditions rapidly differentiate hESCs, and placental marker expression can be detected as early as differentiation day 3.
Transfections of siRNAs and Plasmid DNA and Viral Infections Using In Vitro-derived Trophoblastic Cells
In vitro derived human trophoblastic cells can be transfected with siRNAs to disrupt the gene expression of either coding or noncoding RNAs. We initiated the differentiation of hESCs using BMP4/A/P, performed two rounds of siRNA transfections (using 75 pmol of siRNAs) on differentiation days 1 and 2, and collected the cells for RNA isolation. Quantitative RT-PCR is an effective method to determine the knockdown efficiency for either coding or noncoding genes. Using two siRNAs complementary to different regions of the long, noncoding RNA lncRHOXF119, we obtained 80% knockdown of lncRHOXF1 transcripts (Figure 3A). Next, we transfected one siRNA (25 pmol) for the protein-coding gene hnRNP U, also on differentiation days 1 and 2. We observed an 80% reduction in hnRNP U transcripts following two consecutive transfections. In vitro differentiated trophoblast progenitor cells can also be used to investigate innate immune responses. We performed infections of differentiation day 2 cells using Sendai virus at an MOI of 1 and collected the cells after 8 hr of viral incubation. Using qRT-PCR, we detected abundant viral protein transcripts (Sev-NP) in infected cells (Figure 3B), indicative of active viral propagation in trophoblastic cells. Importantly, infected cells (using MOI = 1) do not change in appearance and are viable (data not shown). In vitro derived trophoblastic cells can also be transfected with plasmid DNA. We performed transfections using a GFP plasmid and introduced this construct into BMP4/A/P-treated cells on differentiation day 1 and day 3 and imaged for GFP the following day (Figure 4). We typically obtained 30-50% transfection efficiency at 24 hr post-transfection. In conclusion, in vitro derived trophoblastic cells can be utilized for gain- and loss-of-function experiments.
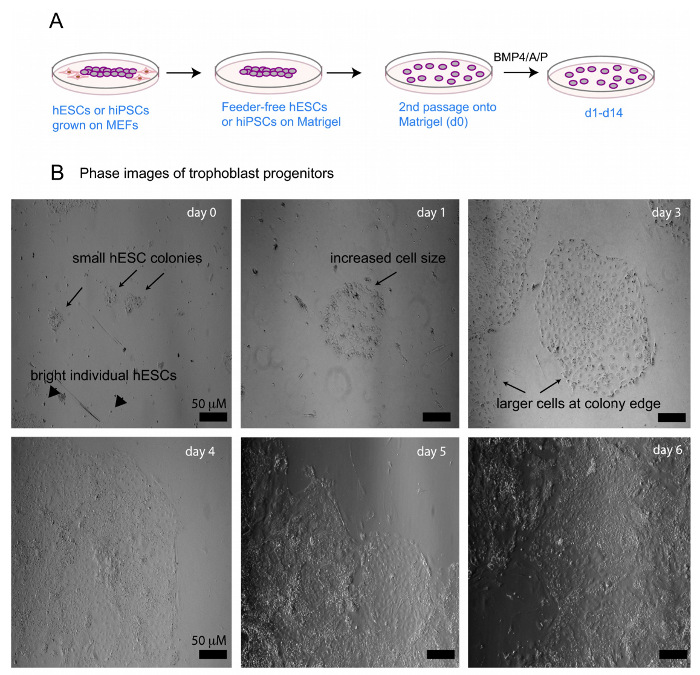
Figure 1: In vitro differentiation of human pluripotent stem cells to early trophoblastic cells using BMP4/A/P. (A) Schematic of BMP4 differentiation of hPSCs to trophoblastic cells using BMP4/A/P. (B) Representative bright-field images of HUES9 cells during d0, d2, and d6 of BMP4/A/P differentiation. Scale bar = 50 μM. The arrowheads denote single-cell hESCs that are not differentiated. The arrows highlight morphological features during differentiation. Please click here to view a larger version of this figure.
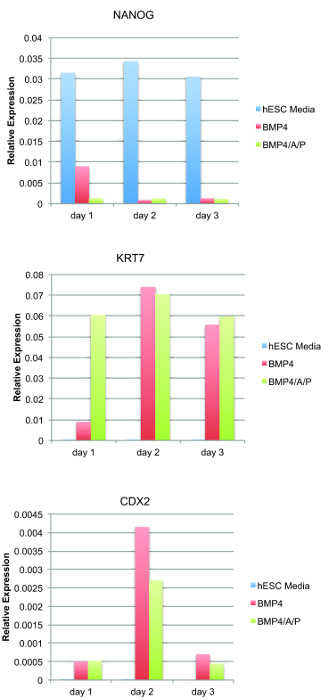
Figure 2: BMP4/A/P differentiation quickly downregulates the pluripotency marker NANOG and upregulates trophoblast genes CDX2 and KRT7. qRT-PCR analysis for NANOG (pluripotency marker), CDX2, and KRT7 (trophoblast markers). The values are averages from triplicate measures. Please click here to view a larger version of this figure.
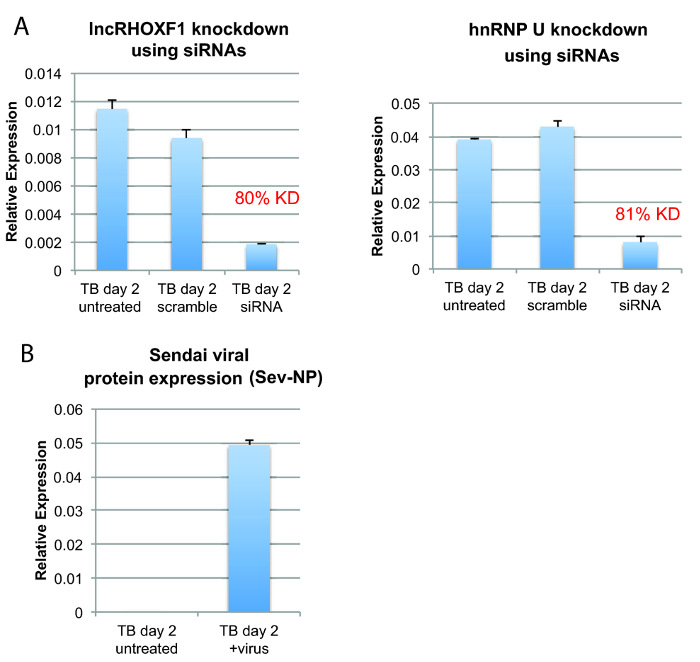
Figure 3: Gene disruption and viral infection using in vitro differentiated human trophoblastic cells. (A) qRT-PCR analysis of hESCs (HUES9) differentiated and then transfected with siRNAs specific to the long, noncoding RNA lncRHOXF1 (left) or the protein-coding gene hnRNP U (right) for two consecutive days. The knockdown efficiency (KD) is typically 70-80%, and the results from one transfection experiment are shown. (B) qRT-PCR analysis of the Sendai viral protein transcript of differentiation day 2 trophoblastic cells infected with Sendai virus (MOI = 1) for 8 hr. The error bars denote the SEM. Please click here to view a larger version of this figure.
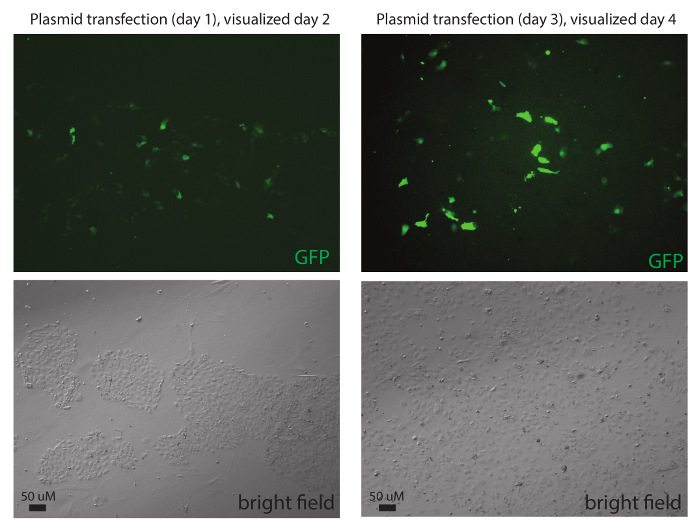
Figure 4: Introduction of plasmid DNA into in vitro differentiated human trophoblastic cells. Transfection of GFP plasmid DNA into differentiation day 1 and day 3 trophectoderm progenitor cells, visualized the following day. Scale bar = 50 μM. Please click here to view a larger version of this figure.
