The structural analysis of proteins and the complexes they form is fundamental for understanding their function. Mass spectrometry considerably contributes to the structural investigation in that it can be applied to almost every complex of interest irrespective of size or sample heterogeneity. We exemplify the protocol by using two well-characterized protein complexes; first, the hetero-dodecamer RvB1/B2 from C. thermophilum and second, the hetero-octameric CPS complex from E. coli.
First, we identified the protein components of the two complexes. For this, the proteins were separated by SDS-PAGE (Figure 2) and gel bands were cut from the gel. After in-gel digestion of the proteins, the peptide mixture was analyzed by liquid chromatography-coupled mass spectrometry and peptide and fragment masses were subjected to database searching. Following this workflow, we identified all protein subunits of the two complexes with high confidence, i.e., a high number of peptides with reasonable peptide scores was observed yielding high sequence coverage for all protein subunits (Table 1).
We then analyzed the intact RvB1/B2 complex by native mass spectrometry (Figure 3A). The mass spectrum revealed two species, one at approximately 8,000 m/z and another species at approximately 11,000-12,000 m/z. The calculated masses for these species correspond to the hexameric (RvB1)3(RvB2)3 ring (approximately 310 kDa) and the dodecameric double-ring (RvB1)6(RvB2)6 (approximately 620 kDa). Both peak series show two populations; these originate from a mixture of His-tagged and untagged RvB2 subunits in the complexes. A crystal structure for the RvB1/B2 complex was previously obtained36 and shows the arrangement of the double-ring (Figure 3B). The native mass spectrum therefore confirms the stoichiometry of the intact dodecamer and furthermore reveals a stable sub-complex. In addition, co-existing populations are identified.
To identify protein interaction sites in the RvB1/B2 complex, we chemically cross-linked the purified complex with BS3 cross-linker. We first titrated the amount of BS3 during the cross-linking reaction to determine the optimal concentration. BS3 is amine-specific and covalently links lysine side chains as well as the N-termini of proteins. The cross-linking reaction was followed by SDS-PAGE (Figure 2A). The non-cross-linked complex showed both RvB1 and RvB2 subunits. Adding BS3 to the reaction mixture caused covalent linkage of the proteins resulting in protein bands at higher molecular weight. The SDS gel shows that increasing amounts of BS3 yield higher amounts of cross-linked species while non-cross-linked protein subunits are reduced. We then cut the protein bands from the gel and followed the protocol provided above to identify protein interaction sites. An example spectrum of a cross-linked di-peptide is shown (Figure 3C). The spectrum shows y-ion series of both peptides confirming this protein interaction. In total, we obtained 14 protein interactions, including four cross-links between subunits RvB1 and RvB2 and two cross-links between two copies of RvB2 (Table 2). The results from BS3 cross-linking are visualized in an interaction network (Figure 3D) showing intra-molecular interactions as well as interactions between different subunits. Intra-molecular cross-links suggest that both RvB1 and RvB2 subunits fold in a way that N- and C-terminal domains are in close proximity. Note, that intra-molecular interactions cannot be distinguished from inter-molecular interactions of the same subunit in this case. Inter-molecular cross-links between the two subunits were also observed. Of these, two inter-molecular cross-links between RvB1 and RvB2 could be visualized in the structure validating the cross-linking approach. The other inter-molecular cross-links are located in flexible loops which are not included in the crystal structure. We also identified two cross-links in RvB2 containing the same peptide sequences. These cross-links can unambiguously be classified as inter-molecular as they must originate from two copies of the same protein (Figure 3D). Our cross-linking experiments reveal protein interaction sites within the complex but also within the protein subunits providing insights into their structural arrangement that could also be confirmed by the existing crystal structure (Figure 3B).
The second protein complex that we studied was CPS. The native mass spectrum (Figure 4A) revealed three protein complexes between 6,000 and 12,000 m/z. The largest complex of 640 kDa corresponds to the intact hetero-octamer. The smaller complexes represent two sub-complexes; the dimer of the small and large CPS subunits (160 kDa) and a hetero-tetramer containing two copies of each subunit (320 kDa). These sub-complexes deliver first insights into the protein assembly; i.e., the large and small subunits are in direct contact (as revealed by the hetero-dimer) and the tetramer might be a product of two dimers. To gain more information on the structural arrangement in the intact CPS complex, we performed tandem mass spectrometry (MS/MS) of the hetero-octamer and the hetero-tetramer. In both cases, the small subunit dissociated from the precursor suggesting that the small subunit is located in the periphery of the assembly (Figure 4B). Indeed, the small subunit is peripheral in the available crystal structure (Figure 4C)44.
Chemical cross-linking using the BS3 cross-linker was also performed. Using increasing amounts, the covalent linkage of the CPS subunits was enhanced. After digestion of the proteins and analysis of the peptides as described above, many protein interactions within the large subunit and one cross-link in the small subunit were obtained (Table 2). In addition, similar to the RvB1/B2 complex, we found two inter-molecular cross-links between two copies of the large CPS subunit. These cross-links place the two large subunits facing each other at their C-terminal sides. In a previous study, combining structural mass spectrometry and computational modeling35, we identified three additional interactions in the large subunit which most likely originate from the interface of two copies of the large subunit validated by the crystal structure and the obtained model (Figure 4C and Table 2). These interactions allow the arrangement of the CPS core complex consisting of four large subunits. However, no inter-subunit cross-links between the small and the large subunits were observed. By inspecting the available crystal structure (Figure 4C), it becomes apparent that the interaction surface between the tetrameric core of the complex, consisting of the large subunit, and the peripheral small subunits is very small, which might explain the absence of inter-subunit interactions. This is confirmed by native mass spectrometry which showed that the small subunit readily dissociates from the intact complexes most likely due to a small binding interface. Nonetheless, protein interactions in the CPS complex combined from chemical cross-linking and native mass spectrometry allow deducing their structural arrangement (Figure 4D).
Taken together, the combination of native mass spectrometry and chemical cross-linking coupled with mass spectrometric identification of cross-linked peptides, allows reconstitution of the structural arrangement of both example complexes. While chemical cross-linking revealed arrangement of the protein subunits, for instance the interactions between RvB1 and RvB2 or within the tetrameric core of CPS, native mass spectrometry delivered protein stoichiometries of the intact complexes and prevalent subcomplexes. In the case of CPS, for which no inter-molecular interactions between the two subunits could be observed by chemical cross-linking, native mass spectrometry suggests that each large subunit interacts with one small subunit (Figure 4D). Tandem mass spectrometry proposed the peripheral location of the small subunit in the complex and a small interface between both subunits.
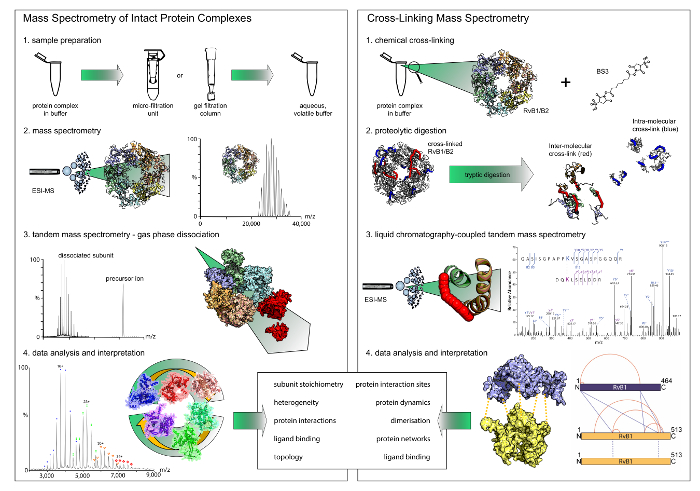
Figure 1: Workflow of native mass spectrometry and cross-linking. Both techniques deliver complementary results. While native mass spectrometry reveals stoichiometries and interaction modules, cross-linking gives insights into the protein interaction sites within the complexes. Note that chemical cross-linking only reveals binary interactions. (A) The first step in native mass spectrometry is buffer exchange to a volatile and aqueous buffer using filter units or gel filtration columns. Mass spectrometry of the intact protein complexes then reveals their stoichiometry. In tandem mass spectrometry experiments, peripheral subunits are dissociated. (B) For chemical cross-linking, the protein complex is incubated with a cross-linking reagent. The cross-linked proteins are then digested into peptides which are subsequently analyzed by liquid chromatography-coupled mass spectrometry. Please click here to view a larger version of this figure.
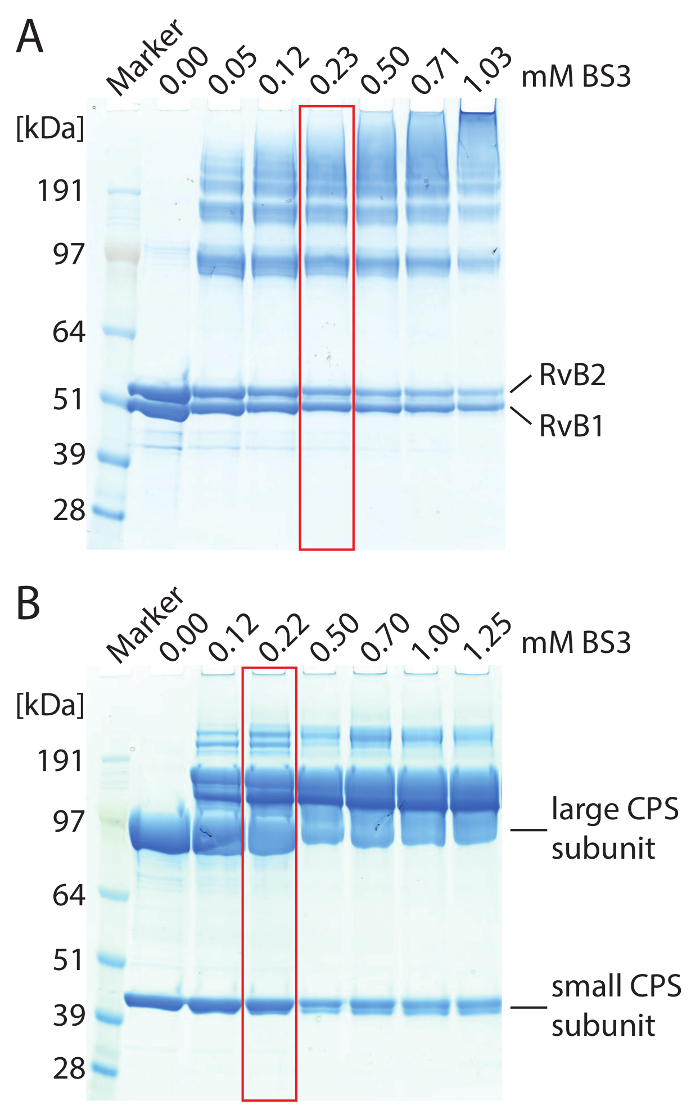
Figure 2: SDS-PAGE of cross-linked RvB1/B2 (A) and CPS (B) complexes. (A) 2.5 µM RvB1/B2 were loaded per gel lane. The concentration of BS3 was varied. Non-cross-linked RvB1/B2 shows the two protein subunits at approximately 50 kDa. Addition of BS3 caused covalent linkage of the protein subunits resulting in protein bands at higher molecular weight. The amount of cross-linked species is increased with higher BS3 concentrations. Optimal cross-linking conditions are highlighted (red). (B) 10 µM CPS were loaded per gel lane. The large (90 kDa) and small (40 kDa) CPS subunits are obtained. Addition of BS3 caused covalent linkage of the protein subunits resulting in protein bands at higher molecular weight. Optimal cross-linking conditions are highlighted (red). Please click here to view a larger version of this figure.
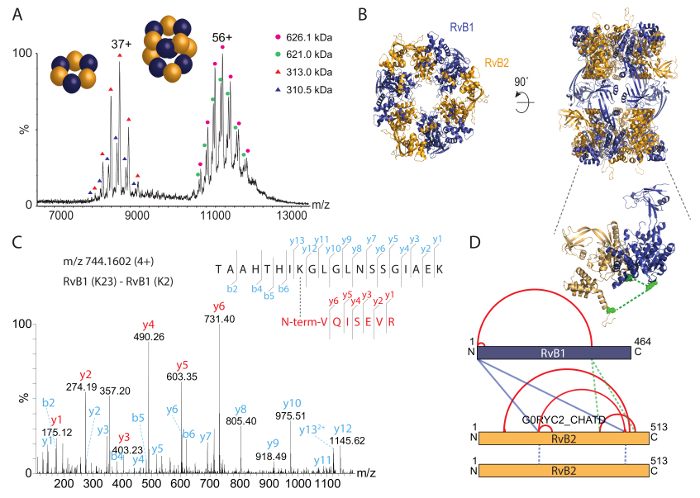
Figure 3: Native mass spectrometry and chemical cross-linking of RvB1/B2 complex. (A) The native mass spectrum reveals two species of RvB1/B2; the intact dodecamer (i.e., (RvB1)6(RvB2)6) at approximately 11,000 to 12,000 m/z and the hexameric ring (RvB1)3(RvB2)3 at approximately 8,000 m/z. Both species show two populations resulting from His-tagged and untagged RvB2. The spectrum has been modified from35. (B) The crystal structure of RvB1/B2 is shown (PDB ID 4WVY). Alternating RvB1 and RvB2 subunits form two hexameric rings. (C) Fragmentation spectrum of a cross-linked di-peptide. The N-terminus of RvB1 was cross-linked with K23 of RvB1. y-ion series were obtained for both peptides (red and cyan). (D) Intra- and inter-protein interactions obtained in the RvB1/B2 complex. Intra-cross-links are shown in red, inter-cross-links are shown in blue. The insert shows two inter-molecular cross-links between RvB1 and RvB2 subunits which could be visualized in the crystal structure (green, insert). Interactions that originate from two RvB2 copies are shown as blue dotted lines. Please click here to view a larger version of this figure.
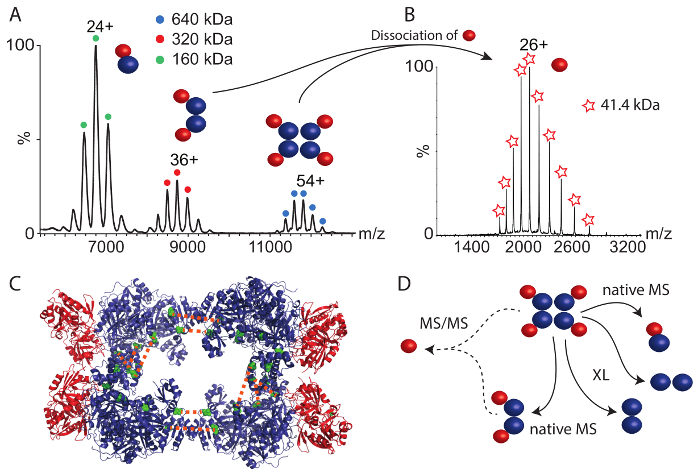
Figure 4: Native mass spectrometry and chemical cross-linking of CPS. (A) The native mass spectrum of CPS shows three complexes. The hetero-dimer (160 kDa), hetero-tetramer (320 kDa) and the hetero-octamer (640 kDa). The spectrum has been modified from35. (B) Tandem mass spectrometry of the tetrameric and octameric CPS complex revealed dissociation of the small CPS subunit. (C) The crystal structure of CPS is shown (PDB ID 1BXR). The large subunits form a tetrameric core and the small subunits are located in the periphery of the complex. Inter-molecular cross-links between two copies of the large subunit are shown (green). (D) Interactions of the large and small CPS subunits. Native mass spectrometry revealed subcomplexes and suggests a peripheral location of the small subunit. Chemical cross-links indicate arrangements in the tetrameric core of CPS. Please click here to view a larger version of this figure.
Table 1: Database search results. The proteins were identified by liquid chromatography-coupled mass spectrometry and database searching. The protein names, accession number, and description as well as protein mass are given. The protein score, number of observed spectra per protein, and the number of observed peptide sequences are listed. The five peptides with highest Mascot peptides scores are listed for each protein subunit. Please click here to download this file.
Table 2: Cross-links observed in RvB1/B2 and CPS. The subunits of the complexes and the cross-linked residues are given. The type of cross-link (intra- or inter-molecular) was revealed from overlapping peptide sequences or a previous study35. Please click here to download this file.
