Motivación
Los esfuerzos para reducir los nutrientes en las aguas superficiales, que son necesarias, entre otras cosas, en el contexto de la aplicación de la Directiva marco del agua Europea1, requieren un examen más detallado de las emisiones de fósforo. El grupo de la sustancia de los fosfonatos (figura 1), que se utilizan como estabilizadores de blanqueo en las industrias textil y de papel, como antiincrustante para tratamiento de agua potable, como estabilizadores de la dureza del agua de enfriamiento y en detergentes y productos de limpieza, es particularmente relevante en términos de cantidad y relevancia ambiental2. Los fosfonatos son sospechosos de contribuir a la eutrofización a largo plazo de cuerpos de agua2,3,4. Por ejemplo, debido a la radiación UV del sol o en presencia de MnII y oxígeno disuelto, los fosfonatos pueden ser degradados en fosfatos microbiológicamente disponibles5,6. El exceso de fosfato es una característica esencial de los cuerpos de agua ecológicamente desequilibrada, que hace el fósforo sustancia objetivo importante para la mejora sostenible de la situación ecológica de los cuerpos de agua.
Fosfonatos pueden eliminarse de las aguas residuales precipitación/floculación cuando usando hierro o aluminio sales7,8,9,10. En este proceso, los metales se transforman en hidróxidos de metal difícilmente solubles. Estos rebaños polar con una relativamente grande superficie específica sirven como adsorbentes para fosfonatos cargados negativamente. Sin embargo, el proceso de floculación puede tener dos inconvenientes principales. Dependiendo de las aguas residuales, volúmenes de lodo de hasta un 30% del volumen de muestra pueden ocurrir11. Este lodo debe ser separados, tratados y eliminados en una etapa de filtro o posterior sedimentación. Además, los fosfonatos pueden complejo los Floculantes añadidos y así evitan la formación de rebaños, sobre todo en aguas con baja dureza. Este efecto puede ser compensado por mayores cantidades de floculante. Sin embargo, esto lleva a valores de mayor β (β = fracción molar de floculante al fósforo en aguas residuales)11,12. Una matriz compleja de aguas residuales, por lo tanto, puede complicar el control de una dosis óptima de floculante.
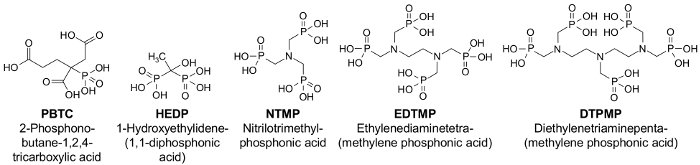
Figura 1: fórmulas estructurales de los fosfonatos importante11. Haga clic aquí para ver una versión más grande de esta figura.
Una posible alternativa que aprovecha la afinidad de adsorción alta de fosfonatos a las superficies que contienen metal y que no tiene el mencionado desventajas son filtro materiales basados en óxidos de hierro (hidr). De dicho material de filtro, la literatura presenta principalmente las investigaciones sobre la eliminación de fosfato13,14,15,16. Este trabajo presenta un procedimiento que permite la investigación de la capacidad de adsorción de los materiales de filtro selectivo de granulado, en este trabajo en particular con hidróxido férrico granular (GFH), con respecto a los fosfonatos con poca carga de trabajo y significativo ahorro de costes. El estudio de la capacidad de adsorción se puede dividir en los siguientes pasos: preparación de la solución de fosfonato, prueba de adsorción (contacto de la solución de fosfonato con el granulado) y análisis de fosfonato. Todas las medidas deberán estar perfectamente coordinadas.
Concepto para la prueba de adsorción y el uso de buffers adecuados
Para el estudio de capacidad de adsorción, pruebas por lotes o columna pueden llevarse a cabo. Para determinar las isotermas de adsorción o pH-dependencias de adsorbente, el enfoque de lote es preferible ya que muchos resultados pueden obtenerse en un período corto de tiempo por la posibilidad de variar varios parámetros. El valor del pH es uno de los factores más importantes que influyen en la adsorción. Cumplimiento o ajuste del valor de pH es un gran desafío para el técnico de laboratorio, como el simple ajuste del valor de pH en la solución de la muestra previamente al contacto con el adsorbente no suele ser suficiente. Cada material adsorbente generalmente se esfuerza por aproximar el pH alrededor de su punto de carga cero (PZC). Por consiguiente, es posible que una solución acuosa, por ejemplo, ajustada a pH 3, cambia a un valor de pH de 8 cuando en contacto inmediato con el adsorbente. Aguas residuales en su mayoría tiene una natural capacidad de almacenamiento en búfer, que atenúa este efecto. Si, sin embargo, sólo la eliminación de una sustancia objetivo particular debe investigarse con un adsorbente particular, se debe utilizar agua residual sintética, es decir, agua pura, que específicamente se enriquecieron con la sustancia objetivo o, por ejemplo, competitivo aniones. Por el contrario adsorbentes a en polvo, donde el valor de pH se puede mantener fácilmente en el rango deseado mediante la adición de ácidos y bases en el recipiente de agitación abierto, sin ajuste de pH en esta forma se pueden hacer en forma de lote con granulados. Para mantener gránulos homogéneamente suspendidos, velocidades de agitación muy elevadas se necesitan, que resultaría en muy rápida abrasión del material. Si tal abrasión es involuntaria, el método suave es girar los tubos de centrífuga cerrada para evitar que los gránulos mezclados continuamente en la solución. La única manera de mantener el pH constante en este caso es usar tampones.
Deben cumplir los siguientes requisitos para los búferes para poder investigar la adsorción de fosfatos y fosfonatos en materiales filtrantes que contienen hierro: libre de fósforo; descolorido; soluble; en el mejor, sin agentes complejantes; no hay competencia con fosfonatos sobre adsorción sobre materiales del filtro polar; estructura similar de los diferentes buffers utilizados; y tampones o sus productos de degradación no deben tener un efecto negativo sobre la absorbancia espectral del color complejo después de la digestión para la determinación de P total. Para el campo de la investigación bioquímica, supuestos buenos almacenadores intermediarios eran desarrollados17,18,19, que tienen exactamente estas propiedades. Así, para las investigaciones de este trabajo, se seleccionaron los buffers en la tabla 1 . El pK el valor cada búfer indica el rango que se puede mantener constante por el búfer. Para el rango de pH < 5, sin embargo, ácidos orgánicos como el ácido cítrico (CitOH) y ácido acético (AcOH) deben usarse. El ácido cítrico es un agente complejante, pero almacena en un rango de pH donde más hierro-que contienen filtro materiales se convierten en inestables de todas formas. Ácido acético y fregonas fueron utilizadas ya por Nowack y piedra7 investigar la adsorción de NTMP sobre Goethita de mezcla (α-FeOOH) a pH 4.6 y 7.2. Sin embargo, sus experimentos en la dependencia de pH de adsorción ocurrieron sin almacenamiento en búfer.
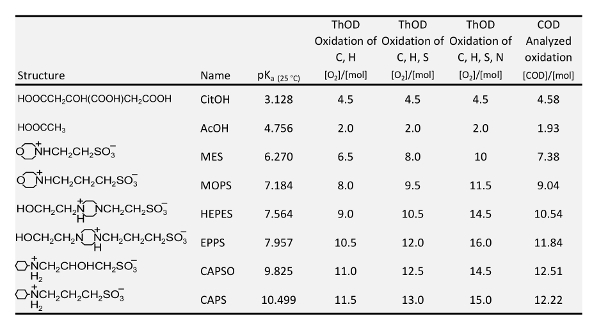
Tabla 1: pK un valores 20 , demanda teórica de oxígeno (DTO) y analizada real demanda química de oxígeno (DQO) de tampones utilizados en este estudio.
Determinación del P total (ISOmini) adaptado a la solución tampón
Después de cada prueba de adsorción, cada solución debe ser analizado para determinar la concentración residual de fosfonato. Recientemente, se introdujo un método para la determinación de los fosfonatos en muestras ambientales con los límites de cuantificación en la gama de 0.1 μg/L. Se basa en el método de IC-ICP-MS y el uso de intercambiadores de cationes (para la conversión de los fosfonatos en ácidos fosfónicos «libres») y aniones intercambiadores de calor (para la concentración previa de los fosfonatos)21. Además, ya en 1997 un método de Nowack22 fue introducido con límites más altos de detección de 15-100 μg/L, que se basa en la pre-complejación de fosfonatos con FeIII, retención con HPLC y detección fotométrica de estos complejos. Sin embargo, estos métodos son muy lentos y costosos. En estudios con aguas residuales sintéticas en las que el único compuesto que contiene fósforo es un fosfonato, es suficiente para determinar la concentración de fosfonato determinando la concentración total de P. La determinación de fosfato inorgánico presenta al experimentador con muchos menos problemas que la determinación del P total, ya que este último requiere digestión previa. La cantidad de químicos que hay que añadir anulan debe coincidir precisamente con los compuestos presentes en la muestra.
La determinación de fosfato en la actualidad se realiza usando principalmente el método introducido por Murphy y Riley23. Este método se basa en la detección espectrofotométrica de un azul intenso color phosphomolybdenum complejo ([PSb2Mo12O40]− con λmax a 880 nm) que se forma en presencia de fosfato y molibdato acidificado con ácido ascórbico y oxidotartrato como agentes reductores24. En otros estudios, la relación óptima de [H+]: [Mo] fue determinada para ser 60-8025,26. Para determinar P total, la digestión, es decir, la ruptura del P-O-P, C-O-P y C P bonos contiene fósforo en compuestos y la oxidación de fósforo fosfato debe llevarse a cabo antes de la formación de azul de phosphomolybdenum24 . EISENREICH et al. 27 presentó un método simplificado basado en el uso del peroxodisulfato de agente oxidante (K2S2O8) en el medio ácido. Muchos de estos hallazgos se han incorporado en el desarrollo de ISO 687828, que explica sistemáticamente el procedimiento para la determinación de fosfato P y concentraciones de P total en muestras de agua (aguas residuales y agua de mar).
La determinación del P total según ISO 6878 (figura 2) requiere la muestra para ser digerido en un matraz de Erlenmeyer por K2S2O8 a un pH ácido (uso de ácido sulfúrico) para por lo menos 30 minutos. Después de la digestión, el valor de pH se establece en 3-10 utilizando NaOH y el contenido de lo Erlenmeyer matraz se transfiere a un matraz aforado de 50 mL. En este frasco, ácido ascórbico y una solución ácida que contiene molibdato y antimonio son añadidos a la muestra y luego con agua. Después de 10-30 minutos, se mide la intensidad de esta coloración azul a una longitud de onda de 880 nm. En el caso de la determinación de fosfato, se omite la digestión. Esto significa que la muestra se mezcla en un matraz aforado de 50 mL con ácido ascórbico y una solución que contiene molibdato así como antimonio, y se mide la intensidad de la coloración azul en el fotómetro.
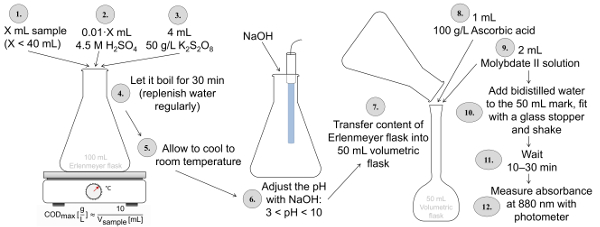
Figura 2 : Procedimiento de determinación del P total según ISO 6878 aplicando digestión con ácido sulfúrico y Potasio peroxodisulfato, un ajuste del pH posterior coloración con ácido ascórbico y que contiene molibdato y NaOH soluciones. Haga clic aquí para ver una versión más grande de esta figura.
El procedimiento de determinación de P total es muy complejo ya que durante la digestión que se debe siempre ser atendido no desborde la muestra y el ajuste de la muestra a pH 3-10 lleva mucho tiempo. Con el fin de ser capaces de analizar tantas muestras como sea posible en un tiempo muy corto, una forma miniaturizada del P total y determinación de Orto fosfato fue desarrollada basándose en este método ISO. Figura 3 resume los pasos individuales de este método. En este método de determinación miniaturizados (ISOmini), el volumen final de la solución de color es de 10 mL (en el método de la ISO, se trata de 50 mL). Por consiguiente, el método demini ISO reduce la cantidad de las soluciones que se utilizarán para la quinta parte. En el método demini ISO, se lleva a cabo la digestión en un termostato (en contraste con el método ISO, donde la digestión se propone en un matraz de Erlenmeyer sobre una plancha) en 148-150 ° C para obtener la mayor oxidación posible. NaOH es añadido después de la digestión junto con la solución ácida de molibdato y ácido ascórbico.
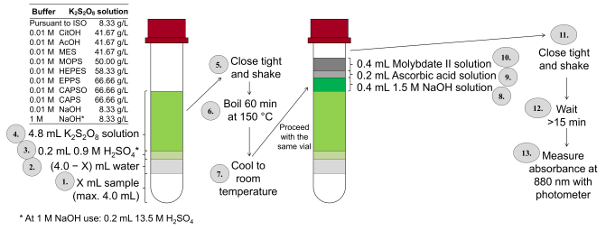
Figura 3 : Procedimiento de determinación del P total según una forma modificada y miniaturizada de ISO 6878 (ISOmini) frascos de uso de tapón de rosca de 10 mL, concentraciones de potasio dependiente del búfer peroxodisulfato, calefacción en un termostato y adición de reactivos a la muestra digerida de color sin la transferencia previamente. Haga clic aquí para ver una versión más grande de esta figura.
Los buffers orgánicos contenidos en las muestras deben estar presentes en concentraciones relativamente altas (10 mM) en comparación con el fosfonato (5-30 μm) para mantener el pH efectivamente. Estos buffers deben ser digeridas para el análisis de la P total después de la prueba de adsorción. Por consiguiente, la cantidad dosificada de agente oxidante debe coincidir con cada memoria intermedia, teniendo en cuenta que demasiado oxidante no debe interferir en la formación del complejo de color formada después de la digestión. Para poder estimar la K2S2O8 cantidad necesaria para la digestión de cada buffer en la determinación de P total basada en el análisis demanda química de oxígeno (DQO), una comparación de cuántos electrones puede convertirse durante el reducción de O2 y K2S2O8 es necesaria:
O2 + 4 H+ + 4 e– → 2 H2O
S2O82 – + 2 e– → 2 así42-
Así, la oxidación de una molécula particular requiere dos veces más moléculas de peroxodisulfato como moléculas de O2 . Por consiguiente, en el caso de un volumen de muestra de 20 mL, el bacalao de la muestra no debe exceder 500 mg/L cuando se utiliza el método ISO. Sin embargo, incluso en el caso del MES, el buen almacenador intermediario con la masa molar más pequeña de la tabla 1, un bacalao de 2,4 g/L está ya presente en una concentración de 10 mM. Además del protocolo paso a paso de la prueba de adsorción y métodomini ISO, este papel, por lo tanto, investiga la concentración del almacenador intermediario necesario, la influencia de los buffers en adsorción de fosfonato y K2S2O8 cantidad y dosis de NaOH requerido para su digestión en el método demini ISO.
Modelo de adsorción de Freundlich
Isotermas de adsorción, es decir, carga q (p. ej., en el adsorbente de P/g de mg) aplicado sobre el c concentración disuelto (en mg/L P) del adsorbente después de un tiempo de contacto específico, pueden ser modelados usando la ecuación propuesta por Freundlich29:
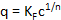
Si los valores obtenidos experimentalmente q y c se trazan en forma de una función ln(q) en ln(c), la pendiente de esta función determinada por regresión lineal corresponde a 1/n y el intercepto del eje y para el valor de KF 30.
Resumen del procedimiento
Todo el proceso para determinar la capacidad de adsorción de hidróxido férrico granular con respecto a los fosfonatos se divide en varios pasos y se describe en la sección de protocolo. Para el análisis, es necesario preparar una cantidad suficiente de reactivo de soluciones (sección 1 del Protocolo). Estas son duraderas durante varias semanas. Luego se prepara la solución de fosfonato que contienen (sección 2), seguido de la prueba de adsorción (contacto de la solución con el material granular de fosfonato) (sección 3) y el análisis de la P total según el método ISO miniaturizado (sección 4).
