Since DNA synthesis is required to incorporate EdU, one can conclude that EdU-labeled nuclei underwent S-phase during the EdU-labeling time window. One may interpret the nuclei that label in a 30 min feeding with EdU labeled bacteria as nuclei in S-phase at the time of dissection. Nuclei that label in a longer continuous EdU feeding experiment may have labeled early in the time window and since left S-phase, or may have labeled in the late part of the EdU time window. EdU signal co-localizes with DAPI signal. In some nuclei, EdU signal covers all chromosomes, while in other nuclei EdU signal localizes to 1–2 bright puncta (Figure 4). These puncta are likely the X-chromosome, which replicates late in S-phase13.
Here, the animals were fed with EdU continuously for 30 min and dissected, as described above and in Figure 5. One example of successful EdU staining in a young adult animal and one example of unsuccessful EdU staining in an older adult animal (see below) are shown in Figure 4. EdU signal from a 30 min labeling localizes to approximately half of the nuclei in the progenitor zone (defined by WAPL-1 antibody labeling but approximated by DAPI morphology26,27,28). S-phase index, the proportion of the progenitor zone that is EdU positive, was previously reported at 57 ±5% and as high as 70% in young adults1,2,3. M-phase index is approximately 2–3%1,29. In continuous feeding for 4 h or longer, all nuclei in the progenitor zone label with EdU, and some nuclei that labeled in the progenitor zone have since entered meiotic prophase1.
While the technique works consistently in wild-type young adult animals, a significant fraction of mated 5 day old hermaphrodites (even those containing sperm) failed to label in a 30 min EdU pulse (Figure 4E). However, with a 4 h EdU feeding, nearly all these animals label. Sporadic failure to label in genetic female animals with short pulses of EdU has also been reported30. There may be other situations that result in sporadic failure to label.
One can calculate the duration of the cell cycle by performing several EdU-labeling experiments with pH3 labeling in each. The duration of G2 was estimated by analyzing the percent of nuclei in M-phase (pH3 immunoreactive) that were EdU positive during the time course (Figure 6). This approach gives median and maximum duration of G2 (Figure 3A). The median time was interpolated, showing an approximate G2 duration of 2.5 h in young adult hermaphrodites. The duration of G2+M+G1 was estimated from the percentage of all progenitor zone nuclei (WAPL-1 immunoreactive) that were EdU positive (Figure 6). The G2+M+G1 method provides a maximum duration measure for the combined phases (Figure 3B). The 99th percentile time was interpolated, showing an approximate G2+M+G1 duration of 3.4 h in young adult hermaphrodites. Data from the same experiments were used to calculate the rate of meiotic entry (nuclei per h). The rate is the slope of the linear regression of the number of nuclei that entered meiosis (EdU positive, WAPL-1 negative or HIM-3 positive) over the duration of the EdU label (Figure 3C). The values for wild-type 1 day old adult hermaphrodites are shown in Table 2.
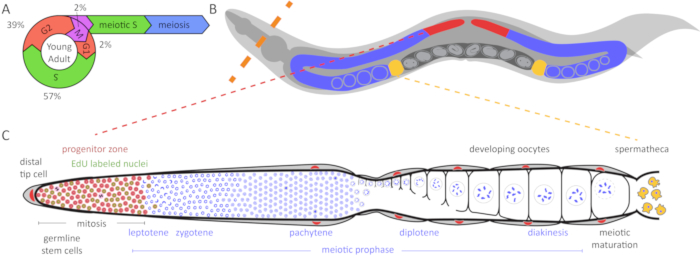
Figure 1: Diagram of C. elegans germline and cell cycle. (A) The cell cycle of germ cells in the young adult hermaphrodite germline. Numbers indicate the approximate percentage of time spent in each cell cycle stage. (B) C. elegans hermaphrodites have two U-shaped germlines (red and blue). The spermatheca is shown in yellow and the uterus with developing embryos is shown in dark gray. The dashed orange line indicates where animals are dissected to extrude the germlines. (C) Diagram of an unfolded C. elegans germline. DAPI (blue) is a DNA dye that highlights nuclear morphology. The distal progenitor zone (highlighted in red based on WAPL-1 antibody staining) contains mitotically cycling stem cells, progenitor cells, and cells in meiotic S-phase (WAPL-1 also labels somatic gonad nuclei). Cells in mitotic and meiotic S-phase label with a 30 min EdU pulse and are indicated in green. Two cells in M-phase label with pH3 antibody and are shown in black. The distal tip cell (DTC) provides the GLP-1/Notch ligand to maintain the stem cell fate of these cells. As the cells migrate away from the DTC, they exit the progenitor zone and enter meiotic prophase. Yellow cells are sperm in the spermatheca. Please click here to view a larger version of this figure.
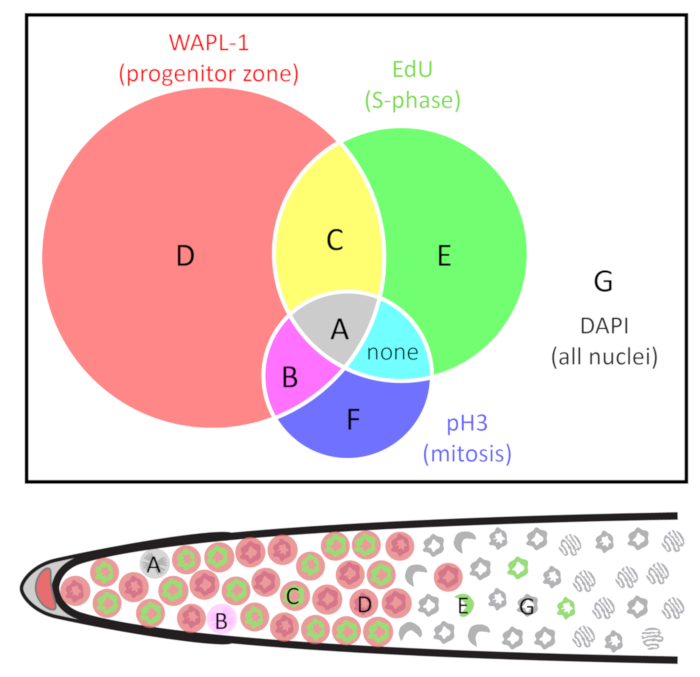
Figure 2: Venn diagram of the classes of nuclei. Nuclei are grouped by the presence and absence of three markers: WAPL-1 indicates progenitor zone cells (red), EdU indicates S-phase cells (green), and pH3 indicates M-phase cells (blue). Cell types are identified as A-G. Note that in wild-type young adult hermaphrodites cells of type F are not found, and cells do not co-label with EdU and pH3 outside of the (WAPL-1 positive) progenitor zone. The distal gonad diagram below indicates one example of A-E and G nuclei. See Table 1 for more detail. Please click here to view a larger version of this figure.
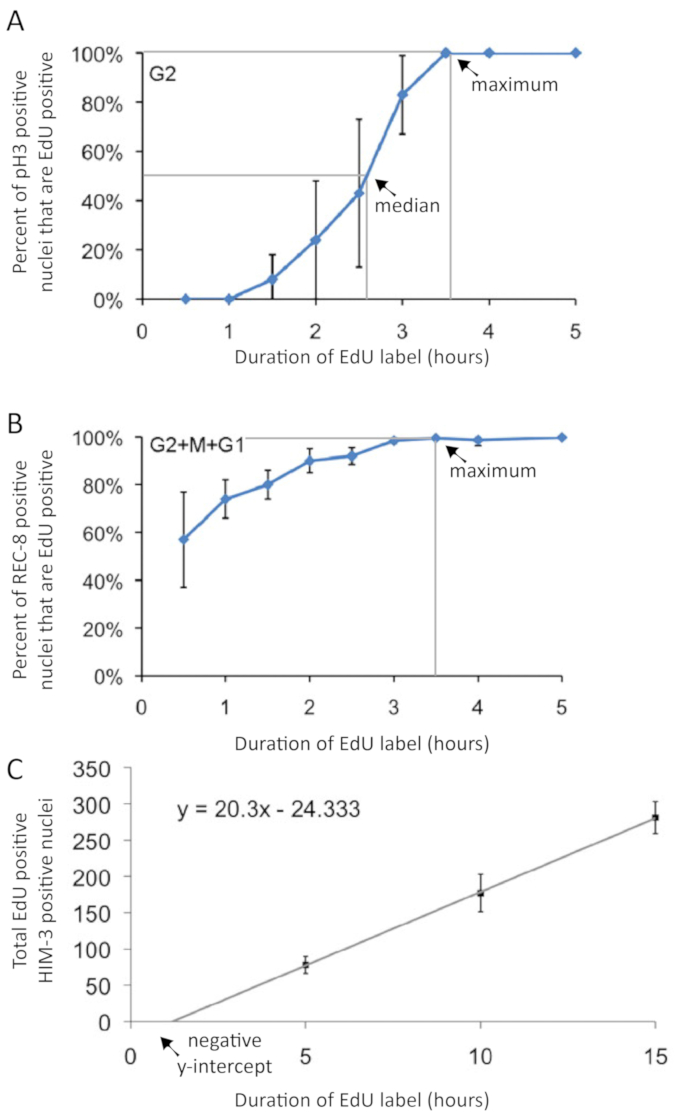
Figure 3: Graphical presentation of cell cycle duration and rate of meiotic entry experimental data. (A) The duration of G2 phase is interpolated from pH3 and EdU co-labeling following varied-duration EdU pulses Gray lines indicate 50th and 99th percentiles used in interpolating median and maximum G2 durations, indicated by arrows. (B) A cell in G2, M, or G1 phase does not incorporate EdU. Thus, the maximum duration of G2+M+G1 phase can be estimated by measuring the maximum duration of EdU label that yields EdU-negative cells. The duration of G2+M+G1 phase is interpolated from EdU and REC-8 co-labeling following varied-duration EdU pulses. Gray line indicates 99th percentile used in interpolating maximum G2+M+G1 duration, indicated by an arrow. It is not possible to interpolate the median G2+M+G1 duration. (C) The rate of meiotic entry (in nuclei per h – see Table 2) is calculated from the slope of the regression line. Note that since the y-intercept intercept is not zero, a regression is necessary for an accurate calculation of the rate of meiotic entry (C). . Error bars indicate standard deviation. Figures modified and reprinted with permission from Fox et al. 20111. Please click here to view a larger version of this figure.
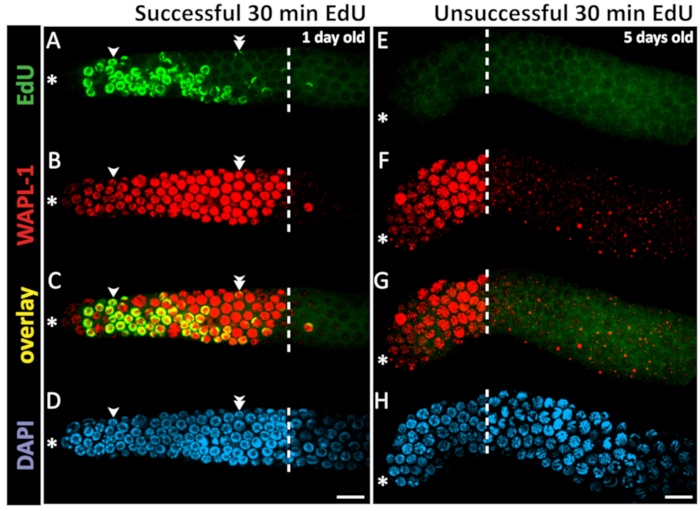
Figure 4: Example of successful and unsuccessful 30 min EdU staining. Confocal microscope images of a 1 day old (A-D) and a 5 day old (E-H) hermaphrodite gonad (not sperm depleted) after a 30 min EdU labeling experiment. The dashed white line marks the end of the progenitor zone. The asterisk marks the position of the distal tip. Green marks EdU staining visualized by click chemistry (A). Unsuccessful EdU labeling results in low-level background staining but no bright EdU+ nuclei (E). Red marks WAPL-1 immunofluorescence (B,F). Yellow indicates overlap (C, G). Blue marks DAPI staining for DNA (D, H). Single arrowheads indicate a nucleus with EdU staining throughout the chromatin. Double arrowheads indicate a nucleus with EdU puncta on only one pair of chromosomes. Images were obtained with a 63X objective. Scale bar = 10 µm (D, H). Please click here to view a larger version of this figure.
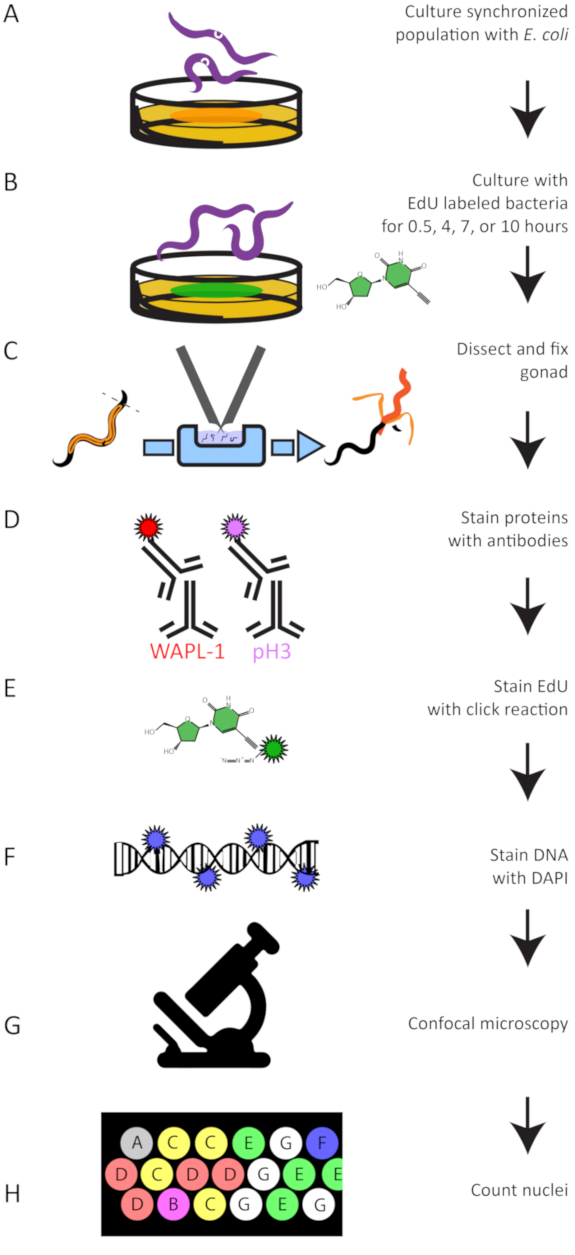
Figure 5: Experimental Workflow. A summary of the experimental protocol to grow (A), EdU label (B), dissect (C), antibody stain (D), perform the click reaction to attach a dye to EdU (E), stain DNA (F), image germlines (G), and quantify EdU labeled and antibody stained nuclei (H). Please click here to view a larger version of this figure.
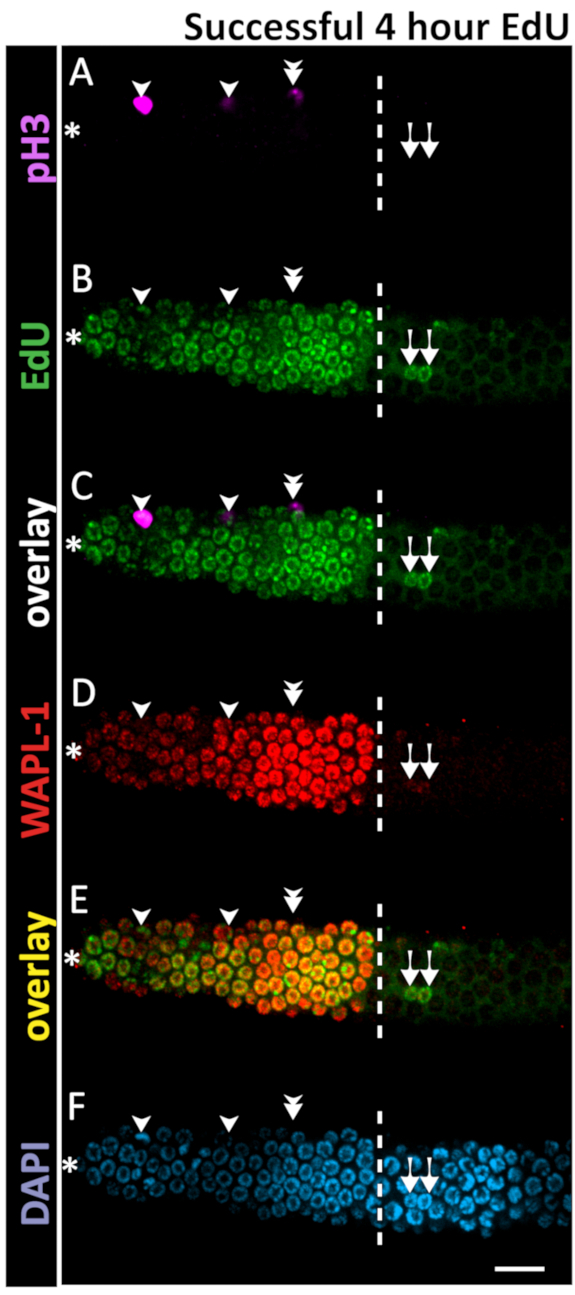
Figure 6: Example of successful 4 h EdU staining. Confocal microscope images of a 1 day old adult hermaphrodite gonad after a 4 h EdU labeling experiment. The dashed white line marks the end of the progenitor zone. The asterisk marks the position of the distal tip. Magenta marks pH3 immunofluorescence (A, C). Green marks EdU staining visualized by click chemistry (B,C). Red marks WAPL-1 immunofluorescence (D). Yellow indicates the overlap of EdU and WAPL-1 (E). Blue marks DAPI staining for DNA (F). Single arrowheads indicate nuclei co-labeled with EdU and pH3. Double arrowhead marks a pH3+ EdU- nucleus – a rare occurrence in a 4 h EdU labeling. Arrows mark EdU+ WAPL-1 – nuclei which have entered meiosis. Images were obtained with a 63X objective. A 10 µm scale bar is shown (F). Please click here to view a larger version of this figure.
| Marker: | pH3 | EdU* | WAPL-1 or REC-8 | HIM-3 | |
| Interpretation: | Mitosis | S-phase* | Progenitor Zone | Meiosis | |
| Class: | Combined Interpretation: | ||||
| A |  |
|
|
 |
in M-phase, in progenitor zone, were in mitotic S-phase during EdU label (completed G2) |
| B | |
|
|
|
in M-phase, in progenitor zone, were not in S-phase during EdU label |
| C | |
|
|
|
in Interphase, in progenitor zone, were in S-phase during EdU label |
| D | |
|
|
|
in Interphase, in progenitor zone, were not in S-phase during EdU label |
| E | |
|
|
|
in meiosis, were in meiotic S-phase during EdU label (meiotic entry nuclei) |
| F | |
|
|
|
return to mitosis (found in some mutants) or meiotic divisions (in spermatogenesis) |
| G | |
|
|
|
in meiosis, were not in S-phase during EdU label |
| sum total pH3 positive; all cells in M-phase | sum total EdU positive; all cells in S-phase | sum total WAPL-1 positive; all cells in progenitor zone | sum total HIM-3 positive; all cells in meiotic prophase |
Table 1: Classes of nuclei. *Note that 30 min and 4 h EdU experiments differ in interpretation. In longer duration EdU experiments, cells have likely progressed beyond S-phase. See Introduction and Step 8 for duration of EdU labeling for relevant experiment.
| Cell cycle part | Operational Definition | Calculation* | Value** |
| Progenitor Zone nuclei | all WAPL-1 (or REC-8) positive, HIM-3 negative nuclei | A+B+C+D | 231 ± 23 nuclei |
| S-phase nuclei | nuclei EdU positive after 30 min EdU label and WAPL-1 positive | A+C | 133 ± 20 nuclei |
| M-phase nuclei | pH3 and WAPL-1 co-positive nuclei | A+B | 5.2 ± 2.3 nuclei |
| S-phase index | S-phase nuclei / Progenitor Zone nuclei | A+C/ A+B+C+D | 57% of cell cycle |
| M-phase index | M-phase nuclei / Progenitor Zone nuclei | A+B/ A+B+C+D | 2% of cell cycle |
| Meiotic Entry cells | EdU labeled nuclei in meiosis | E | varies by duration of EdU label |
| Meiotic Entry rate | Meiotic entry nuclei per h of EdU label | Slope from Figure 4C*** | 20.3 nuclei per h |
| G2 duration (median) | 50% intercept from Figure4A | 2.5 h | |
| G2 duration (maximum) | 99% intercept from Figure 4A | 3.5 h | |
| G2+M+G1 duration (maximum) | 99% intercept from Figure 4B | 3.5 h | |
| Cell cycle duration (median) | median G2 duration / G2-index**** | 6.5 h | |
| Cell cycle duration (maximum) | maximum G2 duration / G2-index**** | 8.1 h | |
Table 2: Cell cycle calculations. *Letters represent the classes of nuclei defined in Table 1 and Figure 3. Calculations are modified from Fox et al. 20111. **Values (± standard deviation) for wild-type hermaphrodites raised at 20 °C aged to 24 h post mid-L4 stage. ***Note that since the y-intercept intercept is not zero, a regression is necessary for an accurate calculation of the rate of meiotic entry. ****The G2-index is determined by subtracting the S-phase index, M-phase index, and approximate G1-index (2%) from 100%, as described by Fox et al. 20111.
