Mono-nucleosomes were first prepared for AFM imaging experiments using a continuous dilution assembly method (Figure 1). The prepared nucleosomes were then checked using discontinuous SDS-PAGE (Figure 2). A mica surface was next functionalized using APS, which captures nucleosomes at the surface while maintaining a smooth background for high-resolution imaging (Figure 3). Nucleosomes were deposited on APS-mica and were subsequently imaged using static AFM imaging. As a control for the assembly and deposition, H3 mono-nucleosomes were prepared and imaged using static AFM. An image of the H3 mono-nucleosomes (Figure 4A) provides a snapshot of the nucleosome population as it existed moments before deposition, confirming that nucleosomes were successfully assembled. The 2 nM nucleosome deposition provided a uniform distribution of nucleosome and DNA particles across the surface and very little to no crowding was observed.
With the H3 control assembly a success, the presented methods were next applied to the study of CENP-A nucleosomes. Static AFM imaging of this sample (Figure 4B) revealed that the assembly was a success. To demonstrate the influence of nucleosome concentration on the surface particle density, the CENP-A nucleosomes were deposited at 1 nM (Figure 4B), compared to the 2 nM used for H3 (Figure 4A). This resulted in a reduced surface particle density for the CENP-A sample to approximately half that of H3 sample. From the static AFM images, the height and turn number of mono-nucleosomes were characterized (Figure 5). Both the angle between the free DNA arms and the length of the free DNA arms was used to determine the number of DNA turns in the individual nucleosome.
Time-lapse AFM imaging of the nucleosomes in buffer was used to visualize the overall spontaneous unwrapping behavior of the nucleosomes (Figure 6). Measuring the angle between nucleosome arms and the contour length of the arms allowed for the turn number to be determined in each of the frames during this unwrapping process (Figure 6 B-C). As the turn number of the nucleosome decreases, a corresponding decrease in the nucleosome core volume is also observed (Figure 6C). High-speed time-lapse AFM was next used to probe the more intricate nucleosome dynamics that were missed using standard time-lapse imaging. The ability of this technique to capture the dynamics over a long period of time was essential to the visualization of a long-distance translocation of a CENP-A nucleosome core (Figure 7) which was captured over the course of ~1200 frames. This technique was also critical in capturing the rare transfer of a CENP-A nucleosome core from one DNA substrate to another (Figure 8). The fast image capture rate (~300 ms/frame) made visualization of this dynamic event possible, as it only took several frames to complete.
Table 1: Reagents needed for continuous salt gradient nucleosome assembly. Each of the components listed is added to the microfuge tube containing the purified DNA. This should be done in the order in which the reagents are listed in the table, with water and NaCl added first, followed by the H2A/H2B dimer and the histone tetramer added last. If pre-folded histone octamers are to be used, add at the same ratio as for the tetramer above. *Take note of the NaCl content in each of the histone stocks and adjust the 5M NaCl to add accordingly, the final [NaCl] should equal 2M. (See Table of Materials).

Figure 1: Schematic of the syringe pump used for microscale nucleosome assembly. The assembly mixture is positioned to be in contact with the end of the syringe needle. As the dilution buffer is delivered by the syringe pump to the assembly mixture the concentration of NaCl is decreased, promoting nucleosome assembly. This figure is adapted from Stumme-Diers et al.30 Please click here to view a larger version of this figure.
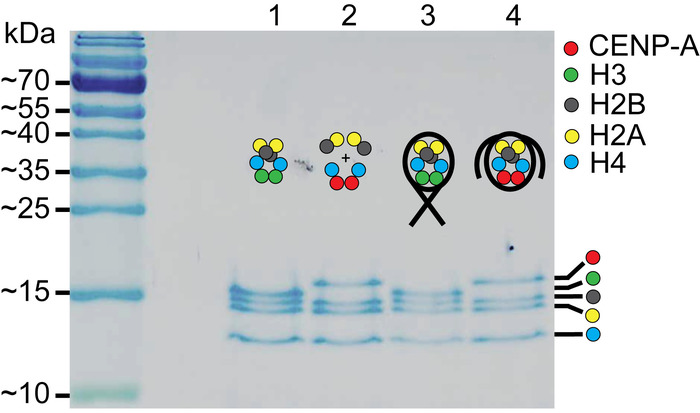
Figure 2: SDS-PAGE of assembled nucleosomes. Lanes 1 and 2 contain the H3 octamer and the CENP-A assembly of histones, respectively. Lanes 3 and 4 contain the assembled H3 nucleosomes and the assembled CENP-A nucleosomes, respectively. Comparison of the assembled nucleosomes to the histone only controls in lanes, confirm that nucleosomes were properly assembled. The cartoon schematic above each lane indicates which histone components are present. This figure is adapted from Stumme-Diers et al.30 Please click here to view a larger version of this figure.
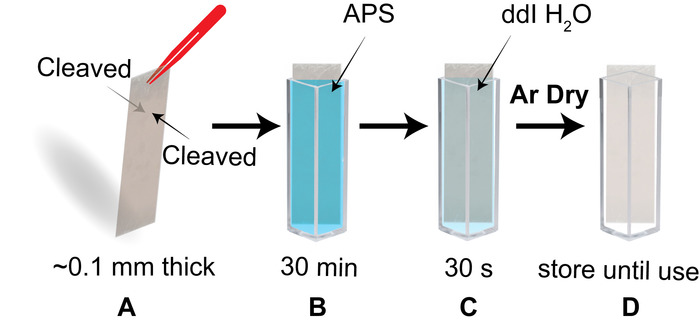
Figure 3: Schematic of the process to prepare APS functionalized mica for AFM imaging of nucleosomes. (A) a piece of mica ~0.1 mm in thickness has both sides freshly cleaved. (B) The cleaved mica piece is promptly placed diagonally in a cuvette containing the APS solution and is set to incubate for 30 min. (C) Following the APS functionalization step, the APS-mica piece is transferred to a cuvette filled with dd H2O for a 30 s rinse. (D) The APS-mica piece is stored in a cuvette until use. Please click here to view a larger version of this figure.
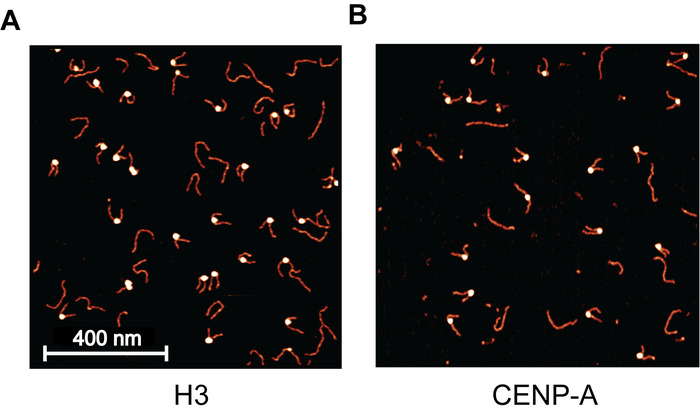
Figure 4: Example AFM images of H3 and CNEP-A nucleosomes. (A) Sample image of H3 mono-nucleosomes deposited on APS-mica, captured using static AFM. Each bright blob is a nucleosome core particle with the flanking DNA regions appearing as noodle-like arms. The long noodle-like features are free DNA particles that are not associated with a histone core. For this image, a 2 nM nucleosome concentration was used, providing a uniform distribution across the surface, with little to no crowding. (B) This nucleosome sample was deposited at 1 nM and is much less populated than the 2 nM used in (A). This demonstrates the direct effect that nucleosome dilution has on the surface density of nucleosomes. Please click here to view a larger version of this figure.
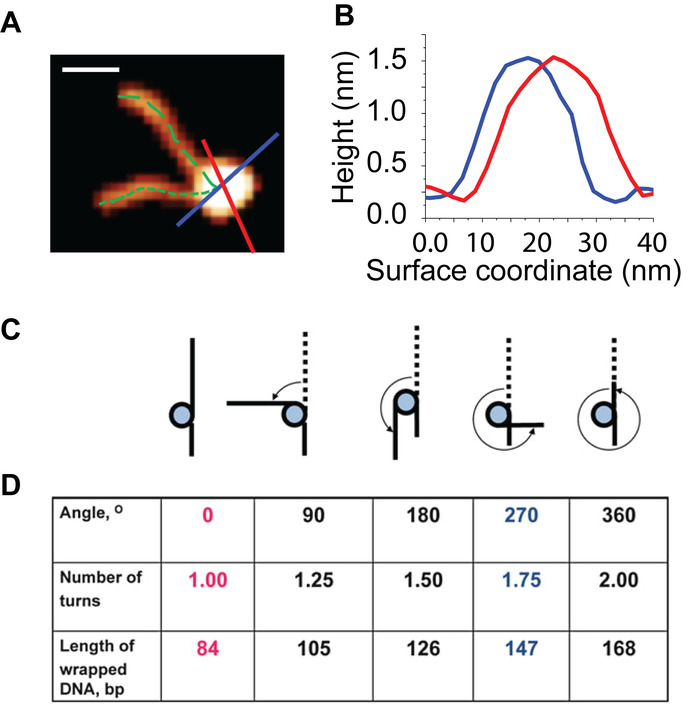
Figure 5: Visual depiction of the analysis used to characterize the wrapping and height of the nucleosome particles. (A) A representative nucleosome particle from images like those shown in Figure 3. The contour length of each nucleosome arm is measured from the end of the arm to the center of the core (dotted green lines). Plotting cross section profiles (red and blue lines) of a nucleosome produce the curves shown in (B) From these curves, height and width detail of particle can be determined. (C) Schematic of the various wrapped states of the nucleosomes. (D) Each wrapped state is characterized using the angle between the DNA arms, the number of DNA turns and the bp of wrapped DNA. Scale Bar = 20 nm. This figure is adapted from Lyubchenko et al. 24 Please click here to view a larger version of this figure.
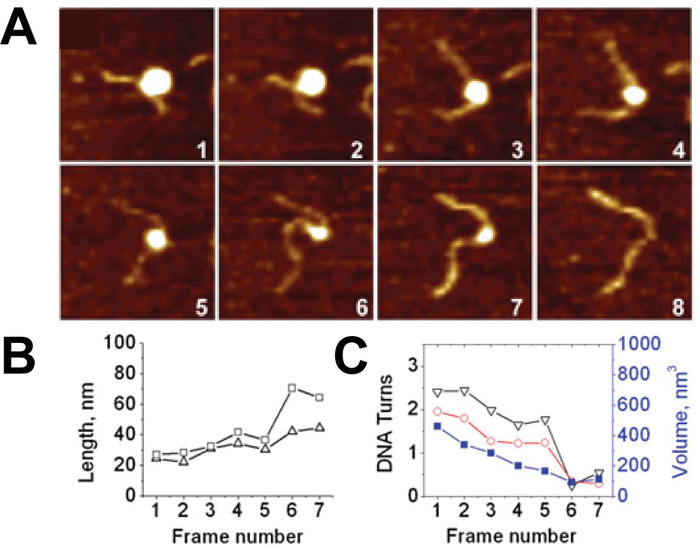
Figure 6. Example of time-lapse AFM images capturing the spontaneous unwrapping of nucleosomes. (A) A series of consecutive AFM images of the spontaneous unwrapping process of nucleosomes captured by continuous scanning in the buffer. The size of each frame is 200 nm and images were captured at a rate of ~170 s per frame. (B) As the unwrapping process progresses in each frame, the arm lengths of the nucleosome increase, (C) resulting in a decrease in DNA turns around the nucleosome. This turn number can be determined from either the measured arm lengths (black) or the angle between the nucleosome arms (red). As the turn number decreases, a reduction in nucleosome volume is also observed (blue curve, right axis). Each frame is 200 x 200 nm in size. This figure is adapted from Lyubchenko et al. 24 Please click here to view a larger version of this figure.
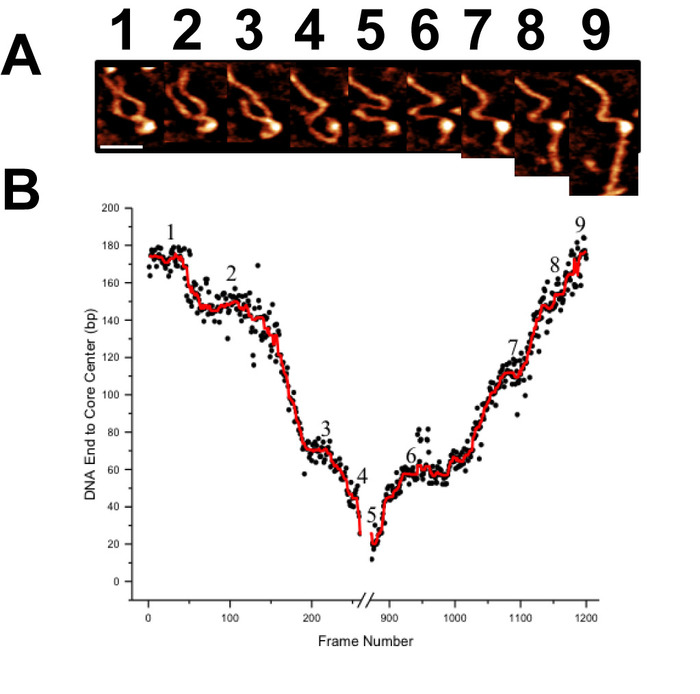
Figure 7. Demonstration of the high capacity of high-speed time-lapse AFM for long image acquisition times (A) A gallery of images selected from more than 1200 frames demonstrating the translocation behavior of a CENP-A nucleosomes core. Each image was captured at a rate of ~300 ms/frame. (B) Contour length measurements from one end of the DNA substrate to the CENP-A core are used to characterize this long translocation process. Scale Bar = 25 nm. This figure is adapted from Stumme-Diers et al. 16 Please click here to view a larger version of this figure.
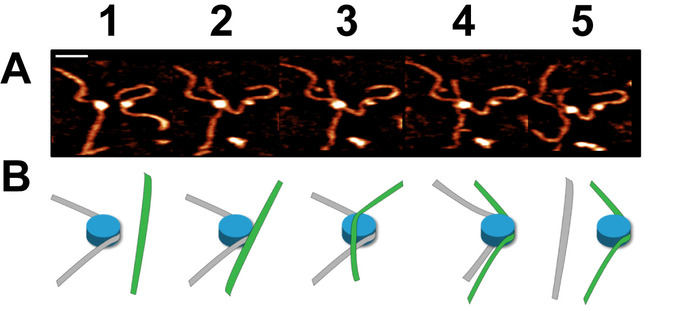
Figure 8. Example of a dynamic nucleosome core transfer captured using high-speed time-lapse AFM (figure adapted from Stumme-Diers et al.16 (A) Selected frames demonstrating the spontaneous transfer of a CENP-A nucleosome core from one DNA substrate to another. This process took place within several frames which were captured at a rate of ~300 ms/frame. (B) A schematic of the transfer process shown in (A). Scale Bar = 25 nm. This figure is adapted from Stumme-Diers et al. 16 Please click here to view a larger version of this figure.
