Agarose cross-linked attivata per lo sviluppo rapido delle resine di maromtagrafia di affinità - Cattura anticorpale come caso di studio
Summary
In questa procedura, un ligando epitope a base di DsRed viene immobilizzato per produrre una resina di affinità altamente selettiva per la cattura di anticorpi monoclonali da estratti vegetali grezzi o supernatanti della coltura cellulare, in alternativa alla Proteina A.
Abstract
La purificazione degli anticorpi monoclonali (mAbs) è comunemente ottenuta dalla cromatografia di affinità Protein A, che può rappresentare fino al 25% dei costi di processo complessivi. Le misure di cattura alternative e convenienti sono quindi preziose per la produzione su scala industriale, in cui vengono prodotte grandi quantità di un singolo mAb. Qui presentiamo un metodo per l’immobilizzazione di un ligando epiteto a base di DsRed a una resina di agarose incrociata che consente la cattura selettiva dell’anticorpo che neutralizza l’HIV 2F5 da estratti vegetali grezzi senza l’uso della proteina A. L’epitopo lineare ELDKWA è stato geneticamente fuso per la proteina fluorescente DsRed e la proteina di fusione è stata espressa in tabacco transgenico (Nicotiana tabacum) prima della purificazione dalla cromatografia dell’affinità glioni-ionive immobilizzata. Inoltre, un metodo basato sull’agarose cross-linked attivato è stato ottimizzato per un’elevata densità di ligandi, accoppiamento efficiente e bassi costi. La composizione del pH e del tampone e la concentrazione di ligando solubile sono stati i parametri più importanti durante la procedura di accoppiamento, che è stata migliorata utilizzando un approccio progettuale degli esperimenti. La resina di affinità risultante è stata testata per la sua capacità di legare selettivamente il mAb target in un estratto di impianto grezzo e il buffer di eluizione è stato ottimizzato per un elevato recupero mAb, attività del prodotto e stabilità della resina di affinità. Il metodo può essere facilmente adattato ad altri anticorpi con epitopi lineari. Le nuove resine consentono condizioni di eluizione più delicate della proteina A e potrebbero anche ridurre i costi di una fase di cattura iniziale per la produzione di mAb.
Introduction
I prodotti biofarmaceutici sono importanti per il trattamento di un ampio spettro di malattie in quasi tutti i rami della medicina1. Gli anticorpi monoclonali (mAbs) dominano il mercato biofarmaceutico, con vendite mondiali che dovrebbero raggiungere quasi 110 miliardi di dollari nel 20202. La piattaforma di espressione favorita per mAbs sono le linee cellulari dell’ovaio del criceto cinese, che in genere producono titer mAb elevati fino a 10 g diL -1 nella coltura supernatant3,4. Tuttavia, la produzione di mAbs nelle colture cellulari dei mammiferi è costosa a causa dell’alto costo del mezzo e della necessità di una fermentazione sterile5. Piattaforme di espressione alternative come gli impianti offrono potenzialmente un approccio più veloce, più semplice, meno costoso e più scalabile per la produzione industriale6,7.
Oltre ai costi associati alle colture cellulari dei mammiferi, l’uso diffuso della cromatografia dell’affinità proteica A per la cattura dei prodotti è un importante fattore di costo per la produzione industriale di mAbs. La proteina A si trova naturalmente sulla superficie delle cellule dello Staphylococcus aureus e si lega alla regione cristallizzabile (Fc) di alcuni anticorpi murini e umani, agendo così come meccanismo di difesa per eludere il sistema immunitario umorale8. La proteina A è diventata il gold standard del settore per la cattura di mAbs dai supernatanti della coltura cellulare ed è anche ampiamente utilizzata dalla comunità di ricerca perché è altamente selettiva, in genere raggiungendo purità mAb del 95% in un solo passaggio8. Non sorprende che le vendite di Protein A negli ultimi due decenni abbiano rispecchiato da vicino le vendite di mAbs8. A seconda della scala di produzione, i costi della proteina A possono corrispondere a più del 25% dei costi totali di processo e quindi influenzare il prezzo di mercato dei mAbs terapeutici, che può essere fino a 2.000 g-15,9. Pertanto, le resine di cromatografia alternativa con prestazioni di purificazione simili hanno il potenziale di ridurre sostanzialmente i costi di produzione, rendendo accessibili terapie basate su anticorpi per un maggior numero di pazienti10,11 ,12. Tali alternative possono anche aggirare gli svantaggi della cromatografia proteica A, comprese le dure condizioni di elusione a basso pH (tipicamente <3.5) che possono potenzialmente causare ai mAbs di subire cambiamenti conformazionali che promuovono l'aggregazione13 . È importante sottolineare che la proteina A è selettiva solo per la regione Fc di alcune sottoclassi IgG, quindi le molecole non funzionali con domini di legame troncati possono co-purificare con il prodotto intatto5, mentre i derivati come i frammenti variabili a catena singola non si legano affatto alla proteina A.
Qui, descriviamo una resina di cromatografia di affinità alternativa per l’acquisizione del mAb 2F5 che neutralizza l’HIV utilizzando il suo epitopo lineare ELDKWA (codice aminoacido di una lettera)5,14. Abbiamo geneticamente fuso l’epitopo 2F5 al capoluogo C della proteina fluorescente DsRed, che funzionava come vettore e molecola reporter, e produceva la proteina risultante DsRed-2F5-Epitope (DFE) nel tabacco transgenico ( Impianti di Nicotiana tabacum. Il DFE è stato purificato dalla cromatografia di affinità agli ioni di metallo immobilizzata in un solo passaggio (IMAC). L’immobilizzazione del ligando di affinità DFE purificato su una resina di agarose cross-linked è stata ottenuta mediante l’accoppiamento chimico utilizzando colonne di agarose ad attivazione n-idrossisuccinimide (NHS). Sono stati poi utilizzati progetti statistici sperimentali per ottimizzare la procedura di immobilizzazione e l’efficienza di accoppiamento15. La strategia di purificazione per mAb 2F5 è stata valutata in termini di purezza, resa e stabilità del ligando degli anticorpi. A differenza della proteina A, che lega la regione fc, DFE legato alla regione di determinazione della complementarità di 2F5, garantendo la purificazione delle molecole con un paratopo intatto. Il nostro concetto può essere facilmente adattato a qualsiasi mAb con un epitopo lineare o ad altri ligandi di affinità basati su peptidi che possono essere facilmente identificati da studi di microarray16.
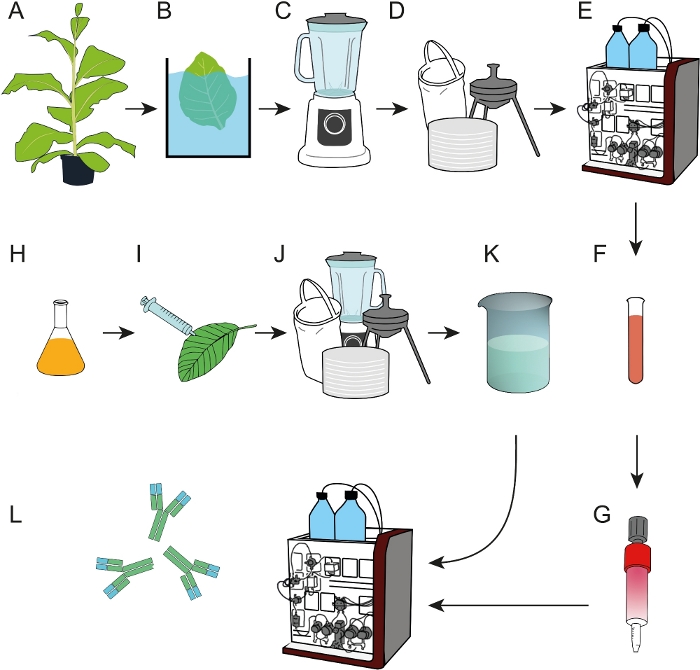
Figura 1: Schema di flusso di processo per la preparazione di resine di affinità epitope che possono essere utilizzate per la cattura di mAbs da estratti vegetali grezzi o supernatanti di coltura cellulare. (A) L’affinità ligando DFE è stata espressa in piante di tabacco transgeniche. Una fase di precipitazione termica (B) è stata inclusa prima dell’omogeneità delle foglie raccolte (C). (D) L’estratto di pianta grezza è stato chiarito dalla filtrazione del sacchetto, dalla filtrazione della profondità e dalla filtrazione sterile di 0,2 m. (E) DFE è stato poi purificato dall’IMAC. (F, G) Il ligando di affinità DFE purificato è stato immobilizzato sulle colonne di agarose incrociate attivate dall’EDC/NHS. (H) Le colture batteriche che trasportano l’anticorpo di codifica T-DNA 2F5 sono state utilizzate per l’espressione transitoria nelle piante di N. benthamiana (I) coltivate in un fitotrone. (J) N. benthamiana foglie sono state raccolte ed elaborate come descritto in D. ( (K) mAb 2F5 è stato purificato dall’estratto chiarificato utilizzando le colonne di affinità DFE (L). Fare clic qui per visualizzare una versione più grande di questa figura.
Protocol
Representative Results
Discussion
Applicazioni della nuova resina di affinità
Abbiamo dimostrato che le resine di cromatografia di affinità personalizzate per la cattura di mAbs possono essere prodotte immobilizzando un ligando contenente un epitopo specifico di mAb all’agarose cross-linked attivata da NHS. Per progettare una tale resina, è stato necessario conoscere la sequenza dell’epitopo e utilizzare un epitopo lineare. Le resine risultanti sono vantaggiose per la cattura di mAbs perché potrebbero potenzialmente sostituire costosi passaggi di cromatografia di affinità proteica A. L’interazione tra 2F5 e DFE nel nostro caso di studio è stata mediata dal legame epitopo-paratopo, quindi il nostro ligando dovrebbe essere più selettivo della proteina A, che si lega alla regione Fc della maggior parte degli IGM murini e umani. Poiché i singoli leganti sono necessari per ogni mAb, il nostro metodo può inizialmente sembrare adatto principalmente per gli anticorpi che vengono prodotti su larga scala. Tuttavia, combinando il nostro approccio con una rapida espressione proteica transitoria a base vegetale, i nuovi ligandi di affinità possono essere preparati in meno di 2 settimane27 con il minimo sforzo28. Quindi, il metodo è adatto anche per la purificazione mAb su piccola scala.
Produzione e potenziali miglioramenti del ligando di affinità
Le piante offrono una piattaforma di produzione veloce e sicura per i ligandi di affinità5,29,30, come la proteina di fusione DFE presente nel nostro case study. Sbiancare il materiale vegetale ha ridotto notevolmente la quantità di proteine delle cellule ospiti in un unico passaggio ed è stato facilmente integrato in una routine di chiarificazione standard. Tuttavia, il recupero del ligando è stato basso nella configurazione attuale, probabilmente a causa della sua moderata stabilità termica e di qualche legame non specifico agli strati del filtro, come riportato per altri prodotti31,32,33. Ingegneria del vettore per aumentare la sua stabilità termica può quindi contribuire a migliorare la resa del ligando in futuro, come descritto per il vaccino antimalarico candidato CCT, l’enzima antitumorPpADI o un glucosidase mesofilo34, 35,36. Lo stesso vale per la fase di filtrazione della profondità, in cui l’ingegneria proteica può contribuire a ridurre il legame non specifico al materiale filtrante37. I costi di produzione per DFE e legature simili potrebbero anche essere ridotti migliorando l’efficienza complessiva del chiarimento utilizzando flocculanti o additivi filtranti38,39.
Quando DsRed viene utilizzato come vettore, forma un complesso tetramerico. Questo è vantaggioso perché aumenta il numero di epitopi per ligando, ma può anche rendere il ligando più suscettibile allo smontaggio o alla denaturazione durante la cromatografia di affinità. Una proteina portante monomerica come mCherry può quindi essere preferibile, perché è stabile a basso pH40, e l’inclusione di ripetizioni tandem dell’epitopo aumenterebbe l’avidità del ligando e quindi aumenterebbe la capacità di resina5, 26 del sistema di , 41.Per le semplici proteine portante-epitopia (cioè quelle senza legami disulfide o modifiche post-traduzionali) i sistemi di produzione microbica possono ridurre i costi di produzione e rendere i ligandi più competitivi con la proteina A. Ad esempio, la proteina fluorescente verde è stata espressa in cellule batteriche con una resa di biomassa di 1g-1 kg, che ridurrebbe significativamente i costi di produzione di ligandi42.
Indipendentemente dall’host dell’espressione, durante l’accoppiamento è stato necessario un ligando di affinità purificato per ridurre al minimo l’immobilizzazione delle proteine cellulari ospiti o dei componenti multimediali che altrimenti possono ridurre la selettività e la capacità della resina. L’inclusione di un tag poli-istidina per la purificazione IMAC ha aumentato la purezza al 90% in un unico passaggio, facilitando la produzione rapida e poco costosa di ligando5,43,44. Tuttavia, la posizione del tag di fusione è importante perché ha il potenziale di ostacolare di tipo sterically il legame dell’epito posta o di indurre la scissione del tag o dell’epitopo dal vettore45,46.
Immobilizzazione del ligando di affinità sulle colonne di cromatografia attivate da NHS
L’immobilizzazione è stata eseguita manualmente o utilizzando un sistema di cromatografia. I piccoli volumi di buffer per colonna sembravano favorire la gestione manuale (ad esempio, a causa dei minimi volumi di rifiuti). Tuttavia, se sono necessarie più colonne o più grandi, il sistema di cromatografia rende le condizioni di accoppiamento più facili da controllare (ad esempio, velocità di flusso regolamentate) ed è quindi più probabile ottenere risultati riproducibili in termini di DBC. I nostri dati suggeriscono che il buffer di accoppiamento e il pH hanno un effetto importante sull’efficienza di accoppiamento e sui costi complessivi delle colonne. I fattori di screening che influenzano la reazione di accoppiamento e la loro regolazione per ogni proteina portante (o anche per ogni fusione carrier-ligando) potrebbero quindi migliorare l’efficienza e le prestazioni della resina di accoppiamento, e consigliamo questo approccio.
Test dell’isolamento 2F5 utilizzando la resina di affinità DFE
La resa e la purezza dei prodotti sono aspetti importanti della resa della resina, e nel caso del DFE abbiamo raggiunto una resa del 105 % e una purezza del 97 % (SD, n. 3), che è paragonabile alle prestazioni delle resine di proteine di riferimentoA 25 ,26. Un altro indicatore chiave delle prestazioni per le resine in generale (e in particolare per quelle basate su ligandi di affinità) è il DBC al 10% di svolta del prodotto, perché questo parametro influisce sulla quantità di resina necessaria per un processo specifico e quindi sui costi. Per il ligando DFE, il DBC iniziale era di 4 g L-1 resina, che è il 13% del valore corrispondente per la Proteina A in condizioni simili (solo 2 min tempo di contatto)25,47 ma circa 15 volte superiore rispetto ad altre resine di affinità personalizzate come la lige anti-FSH-immunoaffinità utilizzando lo stesso tempo di residenza di 2 min48. Il DBC di DFE è sceso al 15% del valore iniziale dopo 25 cicli di legame e di elizione, mentre più di 50 cicli sono necessari per la stessa perdita di DBC nelle resine commercialiA 49. Tuttavia, è importante notare che il nostro vettore non è ancora stato ottimizzato nella stessa misura della proteina A, che è stata studiata e migliorata in modo completo negli ultimi quattro decenni8.
Finora abbiamo migliorato la stabilità della resina e il recupero del prodotto passando da un buffer di eguagliazione a basso pH a un’alta concentrazione di cloruro di magnesio (Figura 3), come raccomandato negli studi precedenti13. Il caratteristico colore rosso del ligando di affinità non è sbiadito sostanzialmente durante i 25 cicli bind-and-elute, quindi ipotizziamo che le proteasi endogene delle piante negli estratti chiarificati della pianta31 possano aver troncato e quindi inattivato l’epitopo di il ligando. Pertanto, la progettazione di linker resistenti alle proteasi per collegare il vettore e l’epitopo può aiutare a mantenere il DBC iniziale su un numero prolungato di cicli. Data la rapida e semplice espressione e purificazione del ligando DFE, il suo semplice accoppiamento alle resine di cromatografia commerciale e la sua eccellente resa e purezza del prodotto, crediamo che il nostro metodo offra un’alternativa adatta alla ProteinA per Purificazione di mAbs e derivati anticorpo che non si legano alla proteina A, soprattutto se i miglioramenti al vettore e al linker possono migliorare la stabilità del DBC e del ligando. Questa ipotesi è stata supportata dalla piccola differenza nella costante di dissociazione dell’anticorpo2F5 2F5 purificato da DFE e dalla proteina A, il che indica che il nostro nuovo ligando di affinità consente il recupero di mAb di alta qualità.
Vantaggi e attuali limitazioni del metodo
La produzione del ligando di affinità come fusione genetica con una proteina portante aumenta la solubilità in tamponi acquosi e quindi la compatibilità con le tipiche condizioni di accoppiamento dei ligandi. Al contrario, i peptidi vuoti derivati dalla sintesi dei peptidi a fase solida possono avere una solubilità limitata in queste condizioni a causa della loro sequenza50, che non può essere modificata perché è dettata dalla sequenza di epitopo di amminoacidi riconosciuta dal mAb essere purificato. Altri hanno quindi utilizzato una sintesi in resina di ligandi peptidi51. La capacità di legatura statica della resina risultante era elevata (80 g di L-1), ma il processo di preparazione della resina è lungo, non è stata riportata una capacità di legame dinamico e la purezza e il recupero ottenuti sono stati inferiori a quelli del nostro approccio. Un ulteriore vantaggio di un ligando di proteine di fusione in scala di laboratorio è che il ligando e le relative varianti possono essere rapidamente prodotti, purificati e testati con il minimo sforzo in un sistema di espressione ad alta velocità di utilizzo52.
Le due attuali limitazioni del metodo qui presentate sono la bassa capacità di legame dinamico di 3 g L-1 e la sua riduzione del 90% nel corso di 25 cicli di legatura-e-elute5. Queste limitazioni possono essere affrontate in futuro applicando condizioni di carico meno rigorose e sostituendo l’attuale supporto DsRed con una variante ingegnerizzata e più stabile rispettivamente. Ad esempio, raddoppiare il tempo di contatto corrente da 2 a 4 minuti ha il potenziale per raddoppiare la capacità di legame dinamico come è stato mostrato per alcune resine proteiche26.
risoluzione f dei problemi
Nella tabella seguente vengono evidenziati i potenziali problemi che possono essere riscontrati durante questo protocollo e vengono forniti suggerimenti su come risolverli (Tabella 1).
| Tabella 1: Potenziali problemi che possono essere riscontrati e possibili correzioni. | |||
| Passaggio del protocollo | problema | Couse | correzione |
| 1 | Le piante non crescono | Condizioni di crescita compromesse | Controllare il pH e la conduttività del fertilizzante |
| Controllare la temperatura e le condizioni della luce | |||
| 2 e 3 | Grandi quantità di proteine delle cellule ospiti sono presenti dopo l’estrazione | Precipitazioni incomplete | Controllare la temperatura durante lo sbollentamento |
| Controllare l’agitazione nel bagno di sbiancamento | |||
| 2 e 3 | Nessun prodotto trovato nell’estratto di pianta | Temperatura di sbiancamento troppo alta | Controllare la temperatura e il pH durante la sbiancamento |
| pH in buffer sbollentamento troppo basso | |||
| 3 | Grandi parti del gambo o della foglia rimangono dopo l’estrazione | Miscelazione incompleta nel frullatore | Assicurarsi che il materiale dell’impianto non formi una spina nel frullatore |
| 3 | Rapido aumento della pressione durante la filtrazione della profondità | Selezione e/o orientamento del filtro non corretti | Controllare il tipo di filtro e l’orientamento |
| 4 | Piccola proteina di fusione durante l’elusione / un sacco di proteina di fusione | La resina IMAC non è stata caricata con ioni metallici | Verificare se la resina IMAC è stata caricata correttamente con ioni |
| La proteina fusione ha perso il tag di affinità | Evitare l’intensa luce solare e le alte temperature durante la coltivazione delle piante | ||
| 4 | Proteina di fusione persa durante la concentrazione | Proteina di fusione legata alla membrana | Controllare il tipo di membrana |
| Assicurarsi che il fattore di concentrazione non sia troppo alto | |||
| 5 | Basso rendimento di accoppiamento | Sequenza errata di aggiunta di reagente di accoppiamento | Controllare le etichette dei reagenti e la sequenza di |
| Preparazione errata delle colonne prima dell’accoppiamento | Controllare le condizioni di colonna preparaiton | ||
| 5 e 6 | Basso rendimento mAb | Bassa espressione mAb nella biomassa vegetale | Testare l’espressione mAb nella biomassa |
| Bassa densità di ligando | Controllare la purezza della preparazione della proteina di fusione | ||
| 7 | Concentrazioni proteiche molto basse/alte nell’ambito di Bradford | Formazione di bolle durante la pipettatura | Controllare le bolle nel palte 96-well |
| 7 | Bassa concentrazione di mAb durante la misurazione SPR | Proteina A compromessa | Confrontare con i risultati di mAb standard con concentrazione nota |
| Diluizione del campione non corretta | Controllare il tasso di diluizione e il buffer | ||
Tabella 1: Risoluzione dei problemi.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Vorremmo riconoscere Ibrahim Al Amedi per aver coltivato le piante di tabacco transgenico e il Dr. Thomas Rademacher per aver fornito il vettore di espressione del tabacco. Gli autori desiderano ringraziare il Dr. Richard M. Twyman per l’assistenza editoriale e Markus Sack per fruttuose discussioni sulla struttura del ligando di affinità DFE. Questo lavoro è stato finanziato in parte dai programmi interni Fraunhofer-Gesellschaft sotto Grant No. Attirare 125-600164 e lo stato del Nord-Reno-Vestfalia sotto il Leistungszentrum grant n. 423 “Produzione in rete e adattiva”. Questo lavoro è stato sostenuto dalla Deutsche Forschungsgemeinschaft (DFG) nel quadro della sovvenzione “Tumor-targeted Drug Delivery” del Gruppo di formazione per la ricerca 331065168. GE healthcare ha sostenuto la pubblicazione ad accesso aperto di questo articolo.
Materials
| 10L/20L Bucket | n/a | n/a | Blanching equipment |
| 2100P Portable Turbidimeter | Hach | 4650000 | Turbidimeter |
| ÄKTApure | GE Helthcare | 29018226 | Chromatography system |
| Allegra 25R | Beckman Coulter | 369434 | Centrifuge |
| Amine Coupling Kit | GE Healthcare | BR100050 | SPR chip coupling kit |
| Amine Coupling Kit | GE Healthcare | BR100050 | SPR chip coupling kit |
| Antibody 2G12 | Fraunhofer IME | n/a | Standard for SPR quantification |
| Blender | Waring | 800EG | Blender |
| BP-410 | Fuhr | 2632410001 | Bag filter |
| CanoScan 5600F | Canon | 2925B009 | Scanner |
| Centrifuge tube 50 mL self-standing | Labomedic | 1110504 | Reaction tube |
| Chelating Sepharose FF | GE Helthcare | 17-0575-01 | Chromatography resin |
| Cond 3320 | WTW | EKA 3338 | Conductometer |
| Design-Expert(R) 8 | Stat-Ease, Inc. | n/a | DoE software |
| Discovery Compfort | Gilson | F81029 | Multichannel pipette |
| Disodium phosphate | Carl Roth GmbH | 4984.3 | Media component |
| Diverse bottles | Schott Duran | n/a | Glas bottles |
| Dri Block DB8 | Techne | Z381373 | Heat block |
| DsRed | Fraunhofe IME | n/a | Standart |
| EDTA | Carl Roth GmbH | 8043.2 | Buffer component |
| EnSpire | Perkin Elmer | 2390-0000 | Plate reader |
| ETHG-912 | Oregon Scientific | 086L001499-230 | Thermometer |
| F9-C | GE Helthcare | 29027743 | Fraction collector |
| Ferty 2 Mega | Kammlott | 5.220072 | Fertilizer |
| Forma -86C ULT freezer | ThermoFisher | 88400 | Freezer |
| HEPES | Carl Roth GmbH | 9105.3 | Buffer component |
| Hettich Centrifuge Mikro 200 | Hettich | 2400 | Centrifuge |
| HiPrep 26/10 | GE Helthcare | GE17-5087-01 | Chromtography column |
| HiTrap NHS-activated Sepharose HP, 1 mL | GE Helthcare | 17-0716-01 | Chromatography columns |
| Hydrochloric acid | Carl Roth GmbH | 4625.1 | Buffer component |
| Imidazole | Carl Roth GmbH | 3899.2 | Buffer component |
| K700 | Pall | 5302305 | Depth filter layer |
| KM02 basic | IKA | n/a | Magnetic stirrer |
| KS50P 60D | Pall | B12486 | Depth filter layer |
| L/S 24 | Masterflex | SN-06508-24 | Tubing |
| Lauda E300 | Lauda Dr Wobser GmbH | Z90010 | Immersion circulator |
| Magnesium chloride | Carl Roth GmbH | KK36.2 | Buffer component |
| Masterflex L/S | Masterflex | HV-77921-75 | Peristaltic pump |
| Minisart 0.2 µm | Sartorius | 16534K | Filter unit |
| Nalgene Rapid-Flow PES bottle top filter | Thermo Fischer Scientific | 595-4520 | Vacuum filtration of SPR buffers |
| Nickel sulphate | Carl Roth GmbH | T111.1 | Buffer component |
| Novex NuPAGE 4-12% BisTris LDS gels | Invitrogen | NP0336BOX | LDS-PAA gels |
| Novex X-cell Mini Cell | Invitrogen | EI0001 | PAGE chamber |
| NuPAGE 20x running buffer | Invitrogen | NP0002 | Buffer concentrate |
| NuPAGE antioxidant | Invitrogen | NP0005 | Antioxidant |
| PageRuler protein ladder (10-180 kDa) | Invitrogen | 26616 | Protein standart |
| Perforated bucked | n/a | n/a | Blanching |
| PH 3110 | WTW | 2AA110 | PH meter |
| PowerPac HC | Biorad | 1645052 | Electrophoresis module |
| Protein A from Staphylococcus aureus | Sigma-Aldrich | P7837-5MG | Coating of SPR chips |
| Sephadex G-25 fine, cross linked dextran | GE Helthcare | 17003301 | Chromatography resin |
| Silicone spoon | n/a | n/a | Spoon |
| Simply Blue SafeStain | Invitrogen | LC6060 | Gel staining solution |
| Sodium acetate | Carl Roth GmbH | 6773.1 | Buffer component |
| Sodium acetate | Carl Roth GmbH | X891.1 | Media component |
| Sodium azide | Sigma Aldrich | S2002-100G | Media component |
| Sodium chloride | Carl Roth GmbH | P029.2 | Buffer component |
| Sodium citrate | Carl Roth GmbH | HN13.2 | Buffer component |
| Sodium bisulfite | Carl Roth GmbH | 243973-100G | Media component |
| Sodium phosophate | Carl Roth GmbH | T877.2 | Media component |
| SPR Affinity Sensor – High Capacity Amine | Sierra Sensors GmbH/Bruker Daltonics | SPR-AS-HCA | SPR chip |
| SPR-2/4 Surface Plasmon Resonance Analyzer | Sierra Sensors GmbH/Bruker Daltonics | n/a | SPR device |
| SSM3 | Stuart | 10034264 | Mini Gyro-rocker |
| Heated vessel, 20 L | Clatronic | n/a | Blanching chamber |
| Sterile syringes, 2 mL | B. Braun | 4606027V | Syringes |
| Syringe adpter (Union Luer F) | GE Helthcare | 181112-51 | Syringe adapter |
| TE6101 | Sartorius | TE6101 | Precision scale |
| Tween-20 (Polysorbate) | Merck | 8170721000 | Buffer component |
| Unicorn 6.4 | GE Helthcare | 29056102 | Chromatography software |
| Vacuum bags | Ikea | 203.392.84 | Plant storge |
| VelaPad 60 | Pall | VP60G03KNH4 | Filter housing |
| Vortex-Genie 2 | Scientific industries | SI-0236 | Vortex |
| XK-26/20 column housing | GE Helthcare | 28-9889-48 | Chromtography column |
References
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnology and Applied Biochemistry. 65 (3), 306-322 (2018).
- Ecker, D. M., Jones, S. D., Levine, H. L. The therapeutic monoclonal antibody market. MAbs. 7 (1), 9-14 (2015).
- Jayapal, K., Wlaschin, K. F., Hu, W. S., Yap, M. G. S. Recombinant Protein Therapeutics from CHO Cells – 20 Years and Counting. Chemical Engineering Progress. 103, 40-47 (2007).
- Kunert, R., Reinhart, D. Advances in recombinant antibody manufacturing. Applied Microbiology and Biotechnology. 100 (8), 3451-3461 (2016).
- Rühl, C., Knödler, M., Opdensteinen, P., Buyel, J. F. A linear epitope coupled to DsRed provides an affinity ligand for the capture of monoclonal antibodies. Journal of Chromatography A. 1571, 55-64 (2018).
- Edgue, G., Twyman, R. M., Beiss, V., Fischer, R., Sack, M. Antibodies from plants for bionanomaterials. Nanomedicine and Nanobiotechnology. 9 (6), e1462 (2017).
- Buyel, J. F., Fischer, R. Very-large-scale production of antibodies in plants: The biologization of manufacturing. Biotechnology Advances. 35 (4), 458-465 (2017).
- Bolton, G. R., Mehta, K. K. The role of more than 40 years of improvement in protein A chromatography in the growth of the therapeutic antibody industry. Biotechnology Progress. 32 (5), 1193-1202 (2016).
- Kelley, B. Industrialization of mAb production technology: the bioprocessing industry at a crossroads. MAbs. 1 (5), 443-452 (2009).
- Brochier, V., Ravault, V. High throughput development of a non protein A monoclonal antibody purification process using mini-columns and bio-layer interferometry. Engineering in Life Sciences. 16 (2), 152-159 (2016).
- Arakawa, T., Futatsumori-Sugai, M., Tsumoto, K., Kita, Y., Sato, H., Ejima, D. MEP HyperCel chromatography II: binding, washing and elution. Protein Expression and Purification. 71 (2), 168-173 (2010).
- Barroso, T. B., Aguiar-Ricardo, R. J., Roque, A. C. Structural evaluation of an alternative Protein A biomimetic ligand for antibody purification. Journal of Computer-aided Molecular Design. 28 (1), 25-34 (2014).
- Mazzer, A. R., Perraud, X., Halley, J., O’Hara, J., Bracewell, D. G. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. Journal of Chromatography A. 1415, 83-90 (2015).
- Sack, M., et al. Functional analysis of the broadly neutralizing human anti-HIV-1 antibody 2F5 produced in transgenic BY-2 suspension cultures. FASEB Journal. 21 (8), 1655-1664 (2007).
- Buyel, J. F., Fischer, R. Characterization of Complex Systems Using the Design of Experiments Approach: Transient Protein Expression in Tobacco as a Case Study. Journal of Visualized Experiments. (83), 51216 (2014).
- Trasatti, J. P., Woo, J., Ladiwala, A., Cramer, S., Karande, P. Rational design of peptide affinity ligands for the purification of therapeutic enzymes. Biotechnology Progress. 34 (4), 987-998 (2018).
- Buyel, J. F., Gruchow, H. M., Boes, A., Fischer, R. Rational design of a host cell protein heat precipitation step simplifies the subsequent purification of recombinant proteins from tobacco. Biochemical Engineering Journal. 88, 162-170 (2014).
- Simonian, M. H., Smith, J. A. Spectrophotometric and colorimetric determination of protein concentration. Current Protocols in Molecular Biology. 76 (Chapter 10), (2006).
- Buyel, F. J., Kaever, T., Buyel, J., Fischer, R. Predictive models for the accumulation of a fluorescent marker protein in tobacco leaves according to the promoter/5’UTR combination. Biotchnology and Bioengeneering. 110 (2), 471-483 (2013).
- Piliarik, M., Vaisocherova, H., Homola, J. Surface plasmon resonance biosensing. Methods in Molecular Biology. 503, 65-88 (2009).
- Buyel, J. F., Fischer, R. Scale-down models to optimize a filter train for the downstream purification of recombinant pharmaceutical proteins produced in tobacco leaves. Biotechnology Journal. 9 (3), 415-425 (2014).
- Baird, G. S., Zacharias, D. A., Tsien, R. Y. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proceedings of the National Academy of Sciences, USA. 97 (22), 11984-11989 (2000).
- Vrzheshch, P. V., Akovbian, N. A., Varfolomeyev, S. D., Verkhusha, V. V. Denaturation and partial renaturation of a tightly tetramerized DsRed protein under mildly acidic conditions. FEBS Letters. 487 (2), 203-208 (2000).
- Firer, M. A. Efficient elution of functional proteins in affinity chromatography. Journal of Biochemical and Biophysical Methods. 49 (1-3), 433-442 (2001).
- Pabst, T. M., et al. Engineering of novel Staphylococcal Protein A ligands to enable milder elution pH and high dynamic binding capacity. Journal of Chromatography A. 1362, 180-185 (2014).
- Müller, E., Vajda, J. Routes to improve binding capacities of affinity resins demonstrated for Protein A chromatography. Journal of Chromatography B. 1021, 159-168 (2016).
- Shamloul, M., Trusa, J., Vadim, M., Vidadi, Y. Optimization and Utilization of Agrobacterium-mediated Transient Protein Production in Nicotiana. Journal of Visualized Experiments. (86), 51204 (2014).
- Rademacher, T., et al. Plant cell packs: a scalable platform for recombinant protein production and metabolic engineering. Plant Biotechnology Journal. , (2018).
- Saxena, L., Iyer, B. K., Ananthanarayan, L. Purification of a bifunctional amylase/protease inhibitor from ragi (Eleusine coracana) by chromatography and its use as an affinity ligand. Journal of Chromatography. B. 878 (19), 1549-1554 (2010).
- Kurppa, K., Reuter, L. J., Ritala, A., Linder, M. B., Joensuu, J. J. In-solution antibody harvesting with a plant-produced hydrophobin-Protein A fusion. Plant Biotechnology Journal. 16 (2), 404-414 (2018).
- Menzel, S., et al. Optimized Blanching Reduces the Host Cell Protein Content and Substantially Enhances the Recovery and Stability of Two Plant-Derived Malaria Vaccine Candidates. Frontiers in Plant Science. 7 (159), 1-7 (2016).
- Yigzaw, Y., Piper, R., Tran, M., Shukla, A. A. Exploitation of the adsorptive properties of depth filters for host cell protein removal during monoclonal antibody purification. Biotechnology Progress. 22 (1), 288-296 (2006).
- Menzel, S., et al. Downstream processing of a plant-derived malaria transmission-blocking vaccine candidate. Protein Expression and Purification. 152, 122-130 (2018).
- Vöpel, N., Boes, A., Edgü, G., Beiss, V. Malaria vaccine candidate antigen targeting the pre-erythrocytic stage of Plasmodium falciparum produced at high level in plants. Biotechnology Journal. 9 (11), 1435-1445 (2014).
- Lee, C. W., Wang, H. J., Hwang, J. K., Tseng, C. P. Protein thermal stability enhancement by designing salt bridges: a combined computational and experimental study. PLoS ONE. 9 (11), e112751 (2014).
- Zhu, L., Cheng, F., Piatkowski, V., Schwaneberg, U. Protein engineering of the antitumor enzyme PpADI for improved thermal resistance. Chembiochem. 15 (2), 276-283 (2014).
- Khanal, O., et al. Contributions of depth filter components to protein adsorption in bioprocessing. Biotechnology and Bioengineering. 115 (8), 1938-1948 (2018).
- Buyel, J. F., Fischer, R. Downstream processing of biopharmaceutical proteins produced in plants: the pros and cons of flocculants. Bioengineered. 5 (2), 138-142 (2014).
- Buyel, J. F. Procedure to Evaluate the Efficiency of Flocculants for the Removal of Dispersed Particles from Plant Extracts. Journal of Visualized Experiments. (110), e53940 (2016).
- Fink, D., et al. Ubiquitous expression of the monomeric red fluorescent protein mCherry in transgenic mice. Genesis. 48 (12), 723-729 (2010).
- Gagnon, P. Technology trends in antibody purification. Journal of Chromatography A. 1221, 57-70 (2012).
- Figueira, M., Laramée, L., Murrell, J. C., Groleau, D., Míguez, C. Production of green fluorescent protein by the methylotrophic bacterium Methylobacterium extorquens. FEMS Microbiology Letters. 193 (2), 195-200 (2001).
- Bornhorst, J. A., Falke, J. J. Purification of proteins using polyhistidine affinity tags. Methods in enzymology. 326, 245-254 (2000).
- Sainsbury, F., Jutras, P. V., Vorster, J., Goulet, M., Michaud, D. A. Chimeric Affinity Tag for Efficient Expression and Chromatographic Purification of Heterologous Proteins from Plants. Frontiers in Plant Science. 7, 141-141 (2016).
- Krupka, M., et al. The Position of His-Tag in Recombinant OspC and Application of Various Adjuvants Affects the Intensity and Quality of Specific Antibody Response after Immunization of Experimental Mice. PLoS ONE. 11 (2), e0148497 (2016).
- Goel, A., et al. Relative position of the hexahistidine tag effects binding properties of a tumor-associated single-chain Fv construct. Biochimica et Biophysica Acta. 1523 (1), 13-20 (2000).
- Tustian, A. D., et al. Development of a novel affinity chromatography resin for platform purification of bispecific antibodies with modified protein a binding avidity. Biotechnology Progress. , (2018).
- Zandian, M., Jungbauer, A. An immunoaffinity column with a monoclonal antibody as ligand for human follicle stimulating hormone. Journal of Separation Science. 32 (10), 1585-1591 (2009).
- Kelley, B. Very large scale monoclonal antibody purification: the case for conventional unit operations. Biotechnology Progress. 23 (5), 995-1008 (2007).
- Petrou, C., Sarigiannis, Y., S, K. o. u. t. s. o. p. o. u. l. o. s. Ch. 1. Peptide Applications in Biomedicine, Biotechnology and Bioengineering. , 1-21 (2018).
- Menegatti, S., et al. Design of protease-resistant peptide ligands for the purification of antibodies from human plasma. Journal of Chromatography A. 1445, 93-104 (2016).
- Rademacher, T., et al. Plant cell packs: a scalable platform for recombinant protein production and metabolic engineering. Plant Biotechnology Journal. , 1-7 (2019).
