Den Proton overføringsprosessen i porphycenes holder potensielle applikasjoner i utviklingen av molekylær brytere, transistorer og informasjonslagring enheter1,2. Spesielt tautomeri i porphycenes gjennom doble Proton overføringsprosessen har tiltrukket bred interesse innen feltene spektroskopi og photophysics2. Den indre hydrogenatomer av porphycene kan migrere fra en trans isomer til den andre tilsvarende trans isomer gjennom dobbel Proton overføringsprosessen som vist i figur 1. To mekanismer har blitt foreslått for den doble Proton overføringsprosessen: den felles og trinnvis mekanisme3,4. I den felles doble Proton overføringsprosessen, både Proton atomer flytte til overgangen staten synkront i en symmetrisk måte, mens en Proton fullfører overføringen før den andre Proton i en trinnvis prosess. To hydrogenatomer kan overføre samtidig eller trinnvis avhengig av korrelasjons styrke mellom to hydrogenatomer5.
Isotopanrikning substitusjon har blitt brukt til å oppdage de strukturelle egenskapene til molekyler og rangere konstanter av reaksjonen Kinetics6. Enkelt deuterium substitusjon i indre hydrogen av porphycene fører til en asymmetrisk form av molekylet. Hydrogen bindingen kan utvide eller kontrakt på grunn av masse forskjell mellom hydrogen og deuterium atomer. Isotopanrikning substitusjon introduserer en forstyrrelsene i stillaset i porphycene. Spørsmålet melder at om asymmetrisk struktur vil påvirke Proton overføringsprosessen. Limbach og kollegaer rapporterte at utskifting av hydrogen med deuterium vil komprimere både hydrogen obligasjoner, og samarbeidende kopling av to hydrogen obligasjoner i porphycene kan favorisere felles mekanisme7, mens Yoshikawa uttalte deuteration ville gjøre trinnvis mekanismen bidra mer enn den felles mekanismen8. Eksperimentelle teknikker, som Force spektroskopi, har blitt utviklet for å fange tautomeri detaljer i en enkelt porphycene9. Men det er fortsatt utfordrende å bestemme Atomic detaljer om Proton overføre eksperimentelt på grunn av sin forbigående natur.
Teoretiske beregninger og simuleringer kan fungere som komplementære verktøy i Elucidating reaksjons mekanismer av Proton overføring. Blant ulike teoretiske metoder, molekylær dynamikk (MD) simuleringer kan overvåke dynamiske bevegelser av hvert Atom, og har vært mye brukt til å avdekke komplekse mekanismer i kjemiske og enzymatisk reaksjoner. Men vanlige MD simuleringer tendens til å lide av utilstrekkelig prøvetaking problemet, spesielt når høy energi barriere finnes i prosessen av interesse. Derfor har forbedrede Prøvetakings metoder blitt utviklet, som inkluderer overgangs bane sampling10,11, paraply prøvetaking (US)12,13, og integrert tempering prøvetaking (Its)14, 15av dem. Kombinasjon av ulike forbedrede sampling metoder kan ytterligere øke sampling effektivitet16,17,18. For å utnytte den forbedrede prøvetaking algoritmer i simulere kjemiske reaksjoner, har vi implementert selektiv integrert tempering prøvetaking (sitter) metode med Quantum mekaniske og molekylære mekaniske (QM/MM) potensialer nylig19. Den foreslåtte sitter-QM/MM metoden kombinerer fordelene fra begge metodene: den sitter metoden akselererer prøvetaking og kan utforske alle mulige reaksjons kanaler uten forkunnskaper i reaksjons mekanismen, og QM/MM gir mer nøyaktig beskrivelse av obligasjons-og obligasjons prosess, som ikke bare kan bli simulert av MM-metoder. Den implementerte sitter-QM/MM tilnærming har lykkes avdekket felles dobbel Proton overføring, uncorrelated og korrelert trinnvis doble Proton overføre mekanisme i ulike systemer, uten pre-definerende reaksjons koordinater19. For porphycene, den trinnvis men korrelert Proton overføring karakter har blitt rapportert19. Hybrid sitter-QM/MM metoden ble brukt til å undersøke isotopanrikning effekt i porphycene i studien vår, og nedenfor er de detaljerte beskrivelser av algoritmen og protokollen av vår metode.
Vi har implementert sitter metode med hybrid QM/MM potensialer. Den effektive potensialet i sitter ble definert til å omfatte den potensielle energien ved ulike temperaturer med vekting faktorer nk for å dekke større temperaturområder,
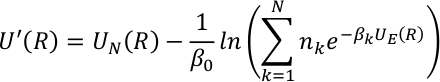
hvor, N er antall kanoniske termer, βk er den inverse temperaturen, og Nk er den tilsvarende vekting faktoren for hver kanoniske komponent. UE (R) og UN(r) representerer de forbedrede og ikke-forbedrede vilkårene i sitter og er definert som,
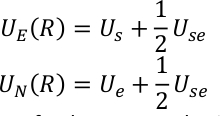
U s, use og Ue er den potensielle energien i sub-system, samspillet mellom sub-system og miljø, og den potensielle energien i miljøet. QM/MM potensial uttrykkes som en hybrid summering av tre komponenter,
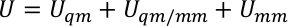
der uQM, uQM/mm, og umm er den interne energien sikt av QM Subsystem, samspillet energien mellom QM og mm regioner, og samspillet energi i mm Subsystem, henholdsvis. UQM/mm sikt kan videre deles inn i tre komponenter, som inkluderer den elektrostatiske, Van der Waals, og kovalente SAMSPILL energi vilkår mellom QM og mm atomer,
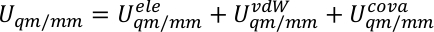
Vi tildeler 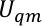 , og
, og 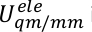 i en Us sikt i sitter,
i en Us sikt i sitter,
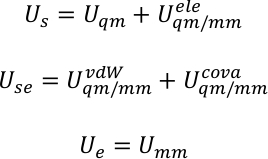
Det fulle potensialet i systemet ble deretter nedbrutt inn i energien i Subsystem us, samspillet energi mellom delsystemet og miljøet use, og energien i miljøet ue. For eksempel, i systemet av dagens arbeid, er delsystemet porphycence, og miljøet vannet.
PMF-profilen langs en kollektiv variabel τ(R) er avledet AS,
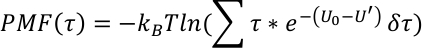
De generelt brukte reaksjons koordinatene for hver hydrogen overføring av N1−H1· · · N2 er q1 = (r1−r2)/2 og q2 = r1 + r2, hvor er1 er avstanden til N1-H1 og r2 er avstanden til H1-N2.
Metoden er implementert i QM/MM MD simulering pakken QM4D20. Den fullstendige kildekoden og dokumentasjonen finner du her: http://www.qm4d.info/.
Generelt, den sitter-QM/MM MD simuleringer innebære fire trinn: pre-likevekt (pre-sitter); optimalisering nk (opt-sitter); produksjons simulering og dataanalyse.
