Descriviamo un approccio di imaging TIRF-N&B (Total Internal Reflection Fluorescence-Number and Brightness) per determinare lo stato oligomerico medio delle molecole recettoriali alla membrana plasmatica delle cellule vive, con l’obiettivo di collegare l’assemblaggio del recettore funzione biologica delle proteine (Figura 1).
Al momento del legame del ligando extracellulare, i recettori avviano la trasduzione del segnale intracellulare a seconda della loro conformazione, oligomerizzazione, potenziali co-recettori e composizione della membrana. Nonostante l’importanza e l’ubiquità dell’oligomerizzazione del recettore, riconosciuto come un evento chiave nella segnalazione cellulare1,2,3,4,5,6, 7, pochi metodi possono rilevare gli eventi di clustering e misurare il grado di clustering sperimentalmente8,9. Il volume confocale (x,y : 300 nm, z ) non è sufficientemente risolto per dimostrare l’interazione molecolare e la stoichiometria, anche dopo l’ottimizzazione da parte degli algoritmi di ripristino delle immagini10. La composizione di sottounità di oligomeri proteici non può essere risolta su base puramente spaziale anche con metodi di super-risoluzione a risoluzione x,y di 20-70 nm come PALM11, STORM12e STED13. Inoltre, la loro risoluzione temporale (nell’ordine dei minuti per immagine) non può seguire la cinetica nell’intervallo di secondi. Lo sbiancamento a passo di singola molecola risolve la stoichiometria degli oligomeri proteici solo se sono immobili14.
Uno dei metodi più versatili per misurare la densità e l’oligomerizzazione delle proteine fluorescenti all’interno di singole immagini è l’analisi della distribuzione dell’intensità spaziale (SpIDA), che si basa sul campionamento spaziale. È applicabile sia alle cellule chimicamente fisse che vive e consente l’analisi di diverse regioni di interesse della cellula contemporaneamente utilizzando la microscopia a fluorescenza standard15. In alternativa, i metodi del momento, come la spettroscopia fluorescenza-correlazione (FCS)16, l’istogramma di conteggio fotone (PCH)17e il numero e la luminosità (N&B)18,19, sono adatti per oligomeri quantitativi Misure. Questi metodi analizzano le fluttuazioni di intensità della fluorescenza che possono essere osservate nel tempo in cui i fluorofori si diffondono dentro e fuori da un volume di illuminazione. Le ampiezze delle fluttuazioni di intensità possono essere descritte in modo univoco dalla luminosità molecolare del fluoroforo e dal numero medio di fluorofori (n) all’interno del volume di illuminazione17 (Figura 2). Tipicamente, il coefficiente di diffusione dei fluorofori e il numero medio di molecole (inversamente correlate al valore G(0) all’interno del volume di illuminazione possono essere ottenuti da FCS20. Tuttavia, poiché il tempo di diffusione si adatta solo alla radice cubica della massa, la FCS non è sufficientemente sensibile per rilevare i cambiamenti nella massa molecolare21. In pratica, fcS monocolore non è in grado di rilevare la dimerizzazione dei recettori della membrana. PCH risolve con precisione le miscele di diversi oligomeri. Utilizzando più di due momenti della distribuzione dell’ampiezza, rileva molecole di luminosità diversa che occupano lo stesso volume di illuminazione. Scansione FCS22 e sviluppi, come l’interessante coppia-correlazione di luminosità molecolare (pCOMB) approccio23, introdotto per estendere la gamma di applicabilità dei metodi di correlazione della fluorescenza nei sistemi biologici24 , rimangono metodi a punto singolo privi della capacità di misurazioni veloci in un’ampia area di una cella, richiedendo molte osservazioni consecutive ad ogni pixel e l’acquisizione di dati nell’ordine di secondi.
N&B è una versione semplificata di PCH che considera solo il primo e il secondo momento dell’ampiezza della distribuzione della fluorescenza, vale a dire l’intensità media, , e la varianza,2 (Figura 2)18,19 e, per questo motivo, non può determinare la frazione molare di oligomeri sconosciuti in una miscela, ma stima solo lo stato medio di oligomerizzazione della miscela. Tuttavia, N&B ha il vantaggio di lavorare con serie temporali relativamente più piccole di immagini di cellule vive rispetto al PCH su base pixel per pixel, semplicemente monitorando le fluttuazioni al momento dell’intensità di fluorescenza. Poiché N&B riduce il tempo per pixel a pochi microsecondi, può seguire cinetiche di oligomerizzazione veloce su grandi aree cellulari, consentendo l’acquisizione di immagini su una scala temporale di secondi nella microscopia a scansione raster (ad esempio, confocale, 2 fotone) e millisecondi microscopia basata su fotocamera (ad es. TIRFM).
Diversi rapporti hanno dimostrato la capacità di N&B di quantificare il numero di sottounità nei cluster proteici mediante l’imaging di regioni cellulari estese. Gli ammassi di Paxillin-EGFP sono stati rilevati nei siti di adesione nelle cellule CHO-K125e l’aggregazione intracellulare del peptide patogeno Httex1p è stata descritta nelle cellule COS-726. N&B è stato richiesto per seguire l’oligomerizzazione guidata dal ligando del recettore ErbB27e l’effetto del ligand FGF21 su Klothob (KLB) e FGFR1c nelle celle HeLa28. La combinazione di imaging TIRF e analisi N&B è stata utilizzata per dimostrare che la dinamina-2 è principalmente tetramerica in tutta la membrana cellulare29. Abbiamo applicato N&B sia alla scansione raster che alle immagini TIRF per dimostrare la dimerizzazione guidata dal ligando dei recettori della membrana cellulare uPAR e FGFR130,31.
I metodi di correlazione della fluorescenza, come N&B, FCS e PCH, si basano sull’idea che in un volume aperto il numero di occupazione delle particelle segua una distribuzione di Poisson. Poiché possono essere rilevati solo i fotoni emettiti dai fluorofori, il valore medio per un’intensità di fluorescenza misurata rispetto al tempo in un pixel dell’immagine,  è il prodotto del numero medio di fluorofori nel volume di illuminazione, n, e la loro luminosità molecolare,17:
è il prodotto del numero medio di fluorofori nel volume di illuminazione, n, e la loro luminosità molecolare,17:

dove il numero di fotoni emessi per unità di tempo (convenzionalmente al secondo) per molecola quando la molecola è al centro del volume di illuminazione.
La luminosità è una proprietà di ogni fluoroforo in una data acquisizione impostata, mentre l’intensità è la somma di tutti i contributi di tutti i fluorofori. Nei concorsi biologici, la luminosità aumenterà con l’aumento del numero di fluorofori che fluttuano insieme, fornendo informazioni sullo stato di oligomerizzazione della proteina fluorescente. Le ampiezze di fluttuazione in un determinato pixel vengono misuratedalla varianza del segnale di fluorescenza,

Dove la media del quadrato  di intensità, , e il
di intensità, , e il  quadrato della media di intensità, , vengono calcolati dai singoli valori di intensità in ogni pixel di ogni fotogramma:
quadrato della media di intensità, , vengono calcolati dai singoli valori di intensità in ogni pixel di ogni fotogramma:
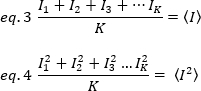
dove K è il numero di fotogrammi totali nella serie temporale. Sperimentalmente, è necessario calcolare per l’intera serie di immagini la varianza che descrive la dispersione dei singoli valori di intensità in ogni pixel di una singola immagine intorno al valore di intensità media. La varianza include tutte le fluttuazioni di origini diverse. In una prima approssimazione, la varianza dovuta alla diffazione delle particelle di fusione nel volume di illuminazione,20, può essere separata dalla varianza a causa del rumore del rivelatore,2d. Le due varianze sono indipendenti; pertanto, la varianza totale è data dalla loro somma:

La varianza, dovuta alle fluttuazioni molecolari all’esterno e all’esterno del volume di rilevamento, dipende linearmente dalla luminosità e dall’intensità molecolari:

Riorganizzazione di eq. 6 in base a eq. 1:

Secondo il concetto tipico nella spettroscopia di correlazione a fluorescenza, l’equazione 7 afferma che la varianza dovuta al numero di fluttuazioni dipende dal quadrato della luminosità della particella.
Quindi, la varianza dovuta alle fluttuazioni del rivelatore è una funzione lineare dell’intensità rilevata, partendo dal presupposto che il rivelatore sia azionato al di sotto del suo limite di saturazione19:

Nel caso dei rilevatori di conteggio dei fotoni unvalore 1 e c.0, quindi la varianza del rivelatore è uguale all’intensità media:

Per applicare questi concetti alle misurazioni reali nelle cellule vive, Gratton e colleghi18 definiscono la luminosità apparente, B, per ogni pixel come rapporto della varianza rispetto all’intensità media:

B è il parametro misurato sperimentalmente. In questo lavoro, le immagini di serie temporali dei recettori FGFR1 nella membrana plasmatica delle cellule HeLa vengono catturate dalla microscopia TIRF e la luminosità media apparente, B, è determinata dall’analisi N&B. Quindi, dopo l’aggiunta di FGF2, serie temporali consecutive vengono catturate per seguire i cambiamenti nell’auto-assemblaggio delle molecole recettoriali nella superficie della membrana dopo la stimolazione del recettore con il ligando canonico.
Tuttavia, poiché il rilevatore del microscopio TIRF è una telecamera EMCCD, l’espressione per la luminosità apparente deve essere modificata come19:

dove offset è l’offset di intensità dell’elettronica di rilevamento che è una caratteristica delle impostazioni del rivelatore. La varianza e l’intensità media per un rilevatore analogico sono date rispettivamente da:
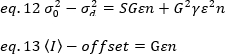
dove G è il guadagno analogico nei livelli digitali (DL/fotoni), S, i livelli digitali per fotone19, è dato dalla pendenza di un’intensità rispetto al grafico di varianza per una fonte di luce con intensità costante (senza fluttuazioni temporali). Il fattore z è correlato alla forma del volume di rilevamento dei pixel. Secondo Hassler et al.32, il fattore z è pari a 0,3 per l’imaging TIRF che lavora al massimo guadagno della telecamera di rilevamento19. I parametri offset, S e G sono caratteristiche della fotocamera e del microscopio. La luminosità apparente, B, si ottiene riorganizzando eq. 11 secondo eq. 12 e 13:

Dal punto di torto dal punto di toesa dal punto di tordo, è una funzione complessa dell’intensità del laser e dell’efficienza di rilevamento del sistema. Tuttavia, dal momento che B/S dipende linearmente da , è importante solo determinare il valore relativo di , per una determinata modalità di rilevamento:

dove è proporzionale a ” s” . Tuttavia, una calibrazione viene eseguita utilizzando un riferimento interno.
