I de siste to tiårene har faste stoffer og filmer som er laget av aggregerte organiske molekyler funnet flere applikasjoner i optoelektroniske enheter. Enheter basert på slike materialer har mange attraktive egenskaper, inkludert liten vekt, fleksibilitet, lavt strømforbruk og potensial for billig produksjon ved hjelp av blekkskriverutskrift. Skjermer basert på organiske lysdioder (OLEDs) erstatter flytende krystallinske skjermer som toppmoderne for mobiltelefoner, bærbare datamaskiner, TV-apparater og andre elektroniske enheter1,,2,,3,,4. Betydningen av OLEDs for belysning applikasjoner forventes å øke i de kommende årene4. Ytelsen til organiske solcelleenheter blir stadig bedre, med effektivitet ved kraftkonvertering over 16 % som nylig ble rapportert for organiske solceller i enkeltkryss5. Organiske materialer har også potensial til å forstyrre andre teknologier, for eksempel fiberoptisk kommunikasjon, hvor deres bruk muliggjør utvikling av elektrooptiske modulatorer med ekstremt høye båndbredder på 15 THz og over6,7.
En stor utfordring med å optimalisere solid-state molekylære materialer for applikasjoner i optoelektronikk er at vanligvis deres egenskaper sterkt avhenger av nanoskala strukturen av materialet. Produksjonsprosessen gjør det mulig å definere nanostrukturen av et materiale til en viss grad ved hjelp av kontrollerte vekstteknikker, for eksempel kjemisk dampdeponering,8 templating av optisk aktive molekyler på et annet materiale (dvs. en polymermatrise9,10), termisk gløding11,12, etc. Nanoskalalidelse er imidlertid iboende til de fleste molekylære materialer og kan vanligvis ikke elimineres helt. Derfor er det viktig å forstå hvordan uorden påvirker egenskapene til et materiale, og å finne måter å konstruere det for optimal ytelse er avgjørende for rasjonell utforming av organiske optoelektroniske materialer.
Graden av lidelse i molekylære materialer er vanligvis for stor til å behandle det som en perturbasjon av en periodisk krystallinsk struktur med en elektronisk struktur som kan beskrives av bandteori. På den annen side er antall molekyler som må inkluderes i en simulering for å reprodusere egenskapene til et bulkmateriale eller en film for stor til å bruke første prinsipper kvantekjemiske metoder som tetthetfunksjonell teori (DFT)13,14 og tidsavhengig tetthet funksjonell teori (TD-DFT)15,16. Organiske molekyler med applikasjoner i optoelektronikk har vanligvis relativt store π-konjugerte systemer; mange har også donor- og aksept- og akseptgrupper. Fange riktig charge-transfer atferd i slike molekyler er avgjørende for å beregne sine optoelektroniske egenskaper, men det kan bare oppnås ved hjelp av langrekkevidde korrigert hybrid funksjonelt i TD-DFT17,,18,19,20. Beregninger som bruker slike funksjonaliseringer skala super lineært med størrelsen på systemet, og i dag er de bare praktiske for modellering av optoelektroniske egenskaper av individuelle organiske molekyler eller små molekylære aggregater som kan beskrives ved hjelp av ikke mer enn ~ 104 atombasis funksjoner. En simuleringsmetode som kan beskrive uordenmaterialer som består av et stort antall kromoforer, vil være svært nyttig for modellering av disse systemene.
Omfanget av intermolekylære interaksjoner i molekylære materialer kan ofte sammenlignes med eller mindre enn rekkefølgen av variasjon i de energiske parametrene (som eigenstate energier eller eksitasjonsenergier) mellom individuelle molekyler som utgjør materialet. I slike tilfeller er multiskala modellering den mest lovende tilnærmingen til å forstå og optimalisere optoelektroniske egenskaper av store uordenmolekylære systemer21,,22,23. Denne tilnærmingen bruker førsteprinsipper kvantekjemiske metoder (vanligvis DFT og TD-DFT) for å nøyaktig beregne egenskapene til individuelle molekyler som komponerer materialet. Hamiltonian av et materiale prøve som er stor nok til å representere bulk molekylært materiale (kanskje, ved å ansette periodiske grenseforhold) blir deretter konstruert ved hjelp av parametrene som ble beregnet for individuelle molekyler. Denne Hamiltonian kan deretter brukes til å beregne optoelektroniske parametere av en stor molekylær aggregat, en tynn film, eller en bulk molekylært materiale.
Exciton modeller er en underklasse av multiskala modeller der spente tilstander av et molekylært materiale er representert på grunnlag av excitons: elektron-hull par som er bundet av Coulomb attraksjon24,25. For modellering av mange spente statlige prosesser er det tilstrekkelig å bare inkludere Frenkel excitons26, hvor elektronet og hullet er lokalisert på samme molekyl. Ladeoverføringseksitoner, der elektronet og hullet er lokalisert på forskjellige molekyler, må kanskje inkluderes i noen tilfeller (f.eks. ved modelleringsladningsseparasjon i donor-acceptor-systemer)27,28. Selv om exciton-modeller er flerskalamodeller som kan parametreres ved hjelp av bare første-prinsippberegninger på individuelle molekyler, står de fortsatt for intermolekylære interaksjoner. De to primære interaksjonstypene de kan ta hensyn til er (a) eksitoniskkoblinger mellom molekyler som karakteriserer excitons evne til å avlokalisere over eller overføre mellom molekyler og (b) elektrostatisk polarisering av elektrontettheten på et molekyl ved ladefordelingen på omkringliggende molekyler. Vi har tidligere vist at begge disse faktorene er viktige for modellering av de optiske og elektrooptiske egenskapene til molekylære aggregater, for eksempel den optiske absorpsjonsspektra29 og første hyperpolarisme30.
I dette papiret presenterer vi en protokoll for parametriserende exciton-modeller som kan brukes til å beregne det optiske spektra og andre optoelektroniske egenskaper av store molekylære aggregater og bulkmolekylære materialer. Den excitonic Hamiltonian antas å være en tett bindende Hamiltonian24,25,
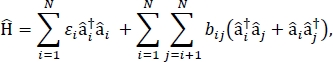
hvor εjeg er eksitasjonenergien til ith molekylet i materialet, bij er den excitonic koblingen mellom ith og jth molekyler, â i† og â jeg er etableringen og utslettelse operatører, henholdsvis for en spent tilstand på ith molekylet i materialet. De excitonic Hamiltonian parametere er funnet ved hjelp av TD-DFT beregninger som utføres på individuelle molekyler som utgjør materialet. I disse TD-DFT-beregningene er ladefordelingen på alle andre molekyler i materialet representert ved elektrostatisk innebygging av atompunktladninger for å ta høyde for elektrostatisk polarisering av et molekyls elektroniske tetthet. Eksitasjonsenergiene, εi, for individuelle molekyler er tatt direkte fra TD-DFT beregningsproduksjonen. De excitonisk koblingene, bij, mellom molekyler beregnes ved hjelp av overgangstetthetskubemetoden31, med bakke-til-spente tilstandsovergangstettheter for de interagerende molekylene tatt fra utgangen av en TD-DFT-beregning i Gaussian32 og etterbehandlet ved hjelp av Multiwfn multifunksjonell bølgefunksjonsanalysator33. For å simulere egenskapene til bulk molekylære faste stoffer, periodiske grenseforhold kan brukes til Hamiltonian.
Den gjeldende protokollen krever at brukeren har tilgang til Gaussian32- og Multiwfn33-programmene. Protokollen har blitt testet ved hjelp av Gaussian 16, revisjon B1 og Multiwfn versjon 3.3.8, men bør også fungere for andre nyere versjoner av disse programmene. I tillegg bruker protokollen et tilpasset C++-verktøy og en rekke tilpassede python 2.7- og Bash-skript, kildekoden som leveres under GNU General Public License (versjon 3) på https://github.com/kocherzhenko/ExcitonicHamiltonian. Beregningene er ment å bli utført på en maskin som kjører et operativsystem fra Unix / Linux-familien.
