This standardized protocol generates a mucociliary epithelial organoid from multipotent progenitors isolated from the early gastrula stage X. laevis embryos within 24 h of cultivation14. Collected deep ectoderm cells self-assemble to form an aggregate in a non-adhesive PCR tube and undergo surface epithelialization and goblet cell differentiation. The newly epithelialized surface of aggregates provides a substrate similar to the native epithelium found in vivo for intercalating inner cells (e.g., multiciliated and other accessory cells) and develops to form mucociliary epithelial organoids (Figure 1A,B). Within 24 h after aggregation, self-organized mucociliary epithelial organoids regenerate a mature epidermis that is indistinguishable from the epidermis of a tadpole. The organoids comprise fully differentiated epithelium (keratin), mucus-secreting goblet cells (ITLN), multiciliated cells (acetylated tubulin), and small secretory cells (peanut agglutinin, PNA) (Figure 1C).
In addition to confirming the development of different cell types with immunostaining, the dynamics of organoid development can be followed by live imaging (Figure 2A). To examine the epithelialization that emerges at the early stage of organoid formation (Figure 1B), we labeled embryos by expressing fluorescently tagged tight junction proteins (ZO-1-RFP) and membrane localizing proteins (mem-GFP). With dual-labeling, the sequential steps of ZO-1-positive tight junction formation can be marked and quantitatively analyzed during epithelialization (Figure 2). For example, for cells (Figure 2B, green-colored) at different stages of epithelialization (at 0 min), some regions of cell-cell adhesion have scattered puncta of ZO-1 (Figure 2B, green arrows). In contrast, other areas have fully assembled, contiguous ZO-1 expression (Figure 2B, yellow arrows). Over time, the puncta coalesce and connect to form contiguous tight junctions (Figure 2B, green arrows), and contiguous tight junctions maintain their morphology even during cell division (Figure 2B, yellow arrows). As the tight junctions mature, cells dynamically move in and out of the surface along the apical planes of the organoids (Figure 2C,D). Furthermore, by tracking cells spatiotemporally on the surface of differentiating organoids (Figure 2B, color-coded cells), multi-scale analysis is possible, ranging from individual puncta to contiguous tight junctions, cell-cell boundaries, and subsets of cell populations within organoids.
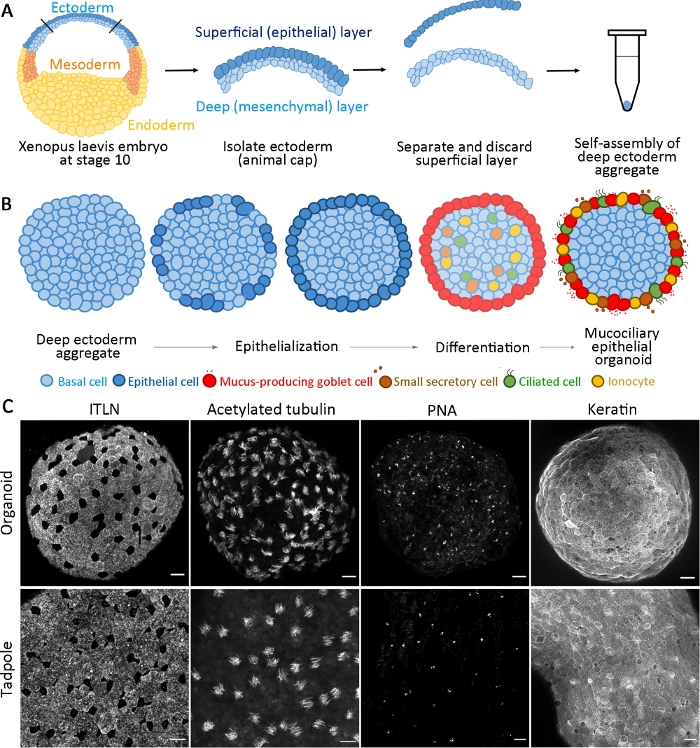
Figure 1: Generation of mucociliary epithelial organoids.
(A) A schematic showing the protocol to assemble deep ectoderm aggregates from X. laevis embryos. (B) A schematic for a model of mucociliary epithelial organoid formation originating from multipotent deep ectoderm cells (cross-sectional view). Surface-positioned cells transit into epithelial cells and differentiate into goblet cells. Differentiating ciliated cells, secretory cells, and ionocytes radially intercalate into the surface and regenerate a mature epidermis. (C) Maximum z-projection of mucociliary epithelium immunostained for ITLN (mucus-producing goblet cells), acetylated tubulin (ciliated cells), PNA (small secretory cells), and keratin (epithelial cells) in organoids at 24 hpa (upper panel) and tadpole epidermis (lower panel). Scale bar = 30 μm. Please click here to view a larger version of this figure.
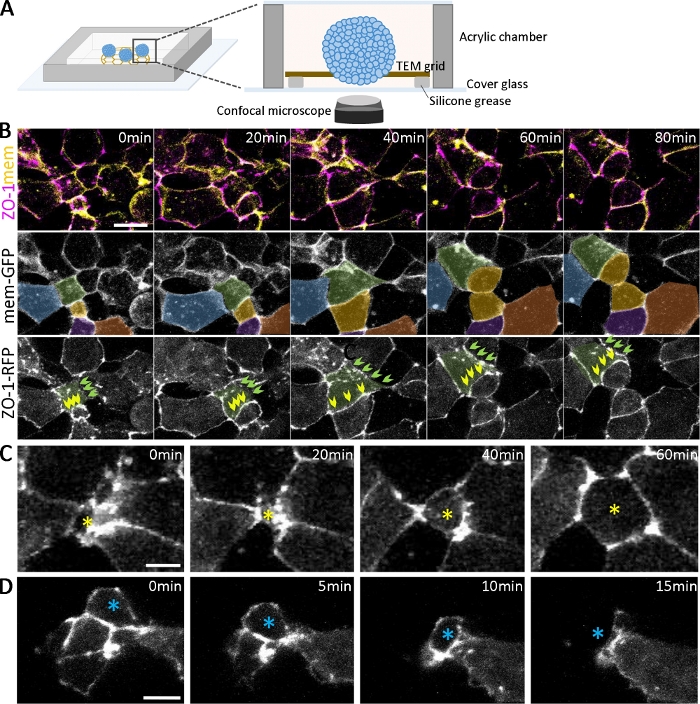
Figure 2: Live imaging of developing organoids.
(A) A schematic of the imaging chamber for live organoids (not to scale). (B) Time-lapse sequences of confocal stacks collected from deep ectoderm cell aggregates expressing ZO-1-RFP and mem-GFP from 2.5 hpa. Scale bar = 20 μm. Cells are pseudo-colored for tracking over time. Green-colored cell have different cell adhesion statuses, including one progressively developing ZO-1 positive adhesion (green arrows) and one maintaining contiguous ZO-1 positive adhesion (yellow arrows) over time. (C, D) Time-lapse confocal images of ZO-1-RFP-expressing deep ectoderm cell aggregates show the radially intercalating cells moving to the surface (C, yellow star) and moving inside the aggregates (D, blue star). Scale bars = 10 μm. Please click here to view a larger version of this figure.
