Quantitative Proteomics Workflow using Multiple Reaction Monitoring Based Detection of Proteins from Human Brain Tissue
Summary
The protocol aims to introduce the use of a triple quadrupole mass spectrometer for Multiple Reaction Monitoring (MRM) of proteins from clinical samples. We have provided a systematic workflow starting from sample preparation to data analysis for clinical samples with all the necessary precautions to be taken.
Abstract
The proteomic analysis of the human brain tissue over the last decade has greatly enhanced our understanding of the brain. However, brain related disorders continue to be a major contributor of deaths around the world, necessitating the need for even greater understanding of their pathobiology. Traditional antibody-based techniques like western blotting or immunohistochemistry suffer from being low-throughput besides being labor-intensive and qualitative or semi-quantitative. Even conventional mass spectrometry-based shotgun approaches fail to provide conclusive evidence to support a certain hypothesis. Targeted proteomics approaches are largely hypothesis driven and differ from the conventional shotgun proteomics approaches that have been long in use. Multiple reaction monitoring is one such targeted approach that requires the use of a special mass spectrometer called the tandem quadrupole mass spectrometer or triple quadrupole mass spectrometer. In the current study, we have systematically highlighted the major steps involved in performing a successful tandem quadrupole mass spectrometry-based proteomics workflow using human brain tissue with an aim to introduce this workflow to a broader research community.
Introduction
During the last decade, rapid developments in mass spectrometry (MS) coupled with increased understanding of chromatography techniques have greatly helped in advancement of MS-based proteomics. Molecular biology-based techniques such as western blotting and immunohistochemistry have long suffered from reproducibility issues, slow turnaround time, inter-observer variability and their inability to accurately quantify proteins, to name a few. To this end, the superior sensitivity of high-throughput proteomics approaches continues to offer molecular biologists an alternate and more reliable tool in their quest to better understand the roles of proteins in cells. However, shotgun proteomics approaches (Data dependent Acquisition or DDA) often fail to detect low abundant proteins in complex tissues besides being heavily reliant on the sensitivity and resolution of the instrument. Over the last couple of years, labs around the world have been developing techniques like Data Independent Acquisition (DIA) that require increased computing power and reliable software that can handle these highly complex datasets. However, these techniques are still a work in progress and not very user friendly. Targeted MS-based proteomics approaches provide a perfect balance between the high throughput nature of MS approaches and the sensitivity of molecular biology approaches like ELISA. A targeted mass spectrometry-based proteomics experiment focuses on detecting hypothesis driven proteins or peptides from discovery-based-shotgun proteomics experiments or through available literature1,2. Multiple Reaction Monitoring (MRM) is one such targeted MS approach that uses a tandem quadrupole mass spectrometer for accurate detection and quantification of proteins/peptides from complex samples. The technique offers higher sensitivity and specificity despite requiring the use of a low-resolution instrument.
A quadrupole is made of 4 parallel rods, with each rod connected to the diagonally opposite rod. A fluctuating field is created between the quadrupole rods by applying alternating RF and DC voltages. The trajectory of the ions inside the quadrupole is influenced by the presence of the same voltages across opposite rods. By applying the RF to DC voltage, the trajectory of the ions can be stabilized. It is this property of the quadrupole that allows it to be used as a mass filter which can selectively let specific ions to pass through. Depending on the need, a quadrupole can be operated in either the static mode or the scanning mode. The static mode allows only ions with a specified m/z to pass through, making the mode highly selective and specific to the ion of interest. The scanning mode on the other hand allows ions across the entire m/z range to pass through. Thus, tandem quadrupole mass spectrometers can operate in 4 possible ways: i) the first quadrupole operating in the static mode while the second operating in scanning mode; ii) the first quadrupole operating in the scanning mode while the second operating in the static mode; iii) both quadrupoles operating in the scanning mode; and iv) both quadrupoles operating in static mode3. In a typical MRM experiment, both the quadrupoles operate in the static mode allowing specific precursors and their resulting products after fragmentation to be monitored. This makes the technique very sensitive and selective allowing accurate quantification.
For molecular biologists, the human brain tissue and its cells are a treasure trove. These remarkable units of an ever-interesting organ of the human body can provide molecular and cellular insights into its functioning. Proteomic investigations of the brain tissue can not only help us understand the systemic functioning of a healthy brain but also the cellular pathways that get dysregulated when inflicted by some disease4. However, the brain tissue with all its heterogeneity is a very complex organ to analyze and requires a concerted approach for a better understanding of the changes at the molecular level. The following work describes the entire workflow starting right from extracting proteins from brain tissue, creating and optimising the methods for MRM assay, to validation of the targets (Figure 1). Here, we have systematically highlighted the major steps involved in a successful MRM based experiment using human brain tissue with an aim to introduce the technique and its challenges to a broader research community.
Protocol
This study involves brain tissue samples from human participants, reviewed and approved by TMH and IITB IEC – (IITB-IEC/2018/019). The participants provided their informed and written consent to participate in this study.
1 Protein extraction from brain tissue
- Weigh around 50 mg of brain tissue and wash the tissue with 300 µL of 1x phosphate buffer saline (PBS) using a micropipette.
NOTE: This step is performed to remove any blood on the external surface of the tissue and must be repeated if necessary. It is advisable to remove as much blood from tissue as possible as it interferes with downstream protein estimation and processing. - Following washes with PBS, add 300 µL of lysis buffer (Buffer A) to the tube containing the tissue. Buffer A contains 8 M urea, 50 mM Tris pH 8.0, 75 mM NaCl, 1 mM MgCl2 and Protease inhibitor cocktail (as per manufacturer's instructions).
NOTE: Protein yield varies depending on several factors ranging from the conditions in which the samples are stored, the amount of starting material and the efficiency of handling the samples while processing. Reduce the volume of buffer A proportionately when working with amounts of tissue lower than 50 mg. - Lyse the tissue using a probe sonicator while keeping the tube on an ice bath. Use the following parameters for sonication: 40% amplitude, 5 seconds on and off cycle for 2:30 min.
- Continue with tissue homogenization using a bead beater to completely lyse the tissue.
NOTE: This step should be performed at medium speed for 90 seconds followed by incubation on ice for 3-5 minutes. - Centrifuge the contents of the tube at 6,000 x g for 15 min at 4 °C. Transfer the supernatant into a fresh tube and mix homogeneously.
NOTE: Make aliquots of the sample and store at -80°C until further use.
2 Protein quantification and quality check
- Quantify the samples prior to digestion using a standard graph made with known concentrations of BSA.
NOTE: Ensure that the assay being used for protein estimation is compatible with the buffer used for making the protein lysate. Check the quality of the protein lysate by running an SDS-PAGE gel.
3 Protein digestion
- Take 50 µg of protein in a micro-centrifuge tube and reduce the proteins by adding Tris 2-carboxyethyl phosphine (TCEP) such that the final concentration is 20 mM. Incubate the contents at 37 °C for 1 h.
- Following incubation, alkylate the reduced proteins by adding iodoacetamide (IAA) to the tube such that its final concentration is 40 mM. Incubate the tube in dark for 30 minutes.
NOTE: Prepare IAA freshly right before its addition to the tube. - Add buffer B to the tube containing the reduced and alkylated proteins such that the final concentration of urea in the tube is less than 1 M.
NOTE: Buffer B is composed of 25 mM Tris (pH 8.0) and 1 mM CaCl2 and is used for diluting the samples. Upon dilution, ensure that the contents of the tube have a pH of 8 for optimum protein digestion after addition of trypsin. - Add trypsin in a 1:30 enzyme to protein ratio and incubate the tubes overnight at 37 °C with constant shaking.
- Following digestion, concentrate the digested product in a vacuum concentrator. At this step, the peptides can either be reconstituted and desalted or stored at -80 °C for future use.
4 Desalting and peptide quantification
NOTE: Desalting or peptide clean-up is essential before loading the samples for LC-MS/MS. Salts and other contaminants in the sample can clog the columns and cause damage to the instrument as well. The process can be performed using commercially available C18 stage-tips or columns.
- Activate the stage tip with 20 µL of 50% acetonitrile (ACN) in 0.1% formic acid (FA). Centrifuge the contents at 1,000 x g for 1 minute and discard the flow through.
NOTE: Conditions for centrifugation are the same till the end of the procedure. - Add 20 µL of 100% ACN in 0.1% FA and centrifuge the contents as in step 4.1.
NOTE: Activation steps can be repeated a couple of times. - Following activation, pass 20 µL of reconstituted peptide sample through the stage-tip and centrifuge as performed in step 4.1.
NOTE: Do not discard the flow through in this step. - Repeat step 4.3 with the flow through at least 5 times to ensure maximal peptide binding to the stage-tip.
- Pass 20 µL of 0.1% FA through the stage-tip and discard the flow through.
NOTE: Repeat this step two more times for better results. - Elute the bound peptides in a fresh microfuge tube by passing increasing concentrations of ACN, i.e., 40%, 60% and 80%, respectively.
- Dry the peptides in a vacuum concentrator and proceed for peptide quantification.
- Reconstitute the dried peptides in 0.1% FA and quantify using the Scopes method5.
5 Transition list preparation of finalized targets
NOTE: A transition refers to the pair of precursors (Q1) to product (Q3) m/z values in an MRM experiment. A peptide can have one to many transitions, with the same Q1 value but different Q3 values. A triple quadrupole mass spectrometer requires information of the transitions for the peptides and their products to be detected. Hence, before starting a targeted experiment, a transition list needs to be prepared. This can be done using the online repository of SRMAtlas6 (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/GetTransitions) or an open source software called Skyline7 (https://skyline.ms/project/home/software/Skyline/begin.view).
- Download the recent human proteome FASTA file from UniProt (https://www.uniprot.org/) and create a background proteome file by inserting it in Skyline. In peptide settings, go to the Background Proteome dropdown and click on Add. Feed in the FASTA sequence file of the human proteome. Make sure this database is selected and the allowed missed cleavage value is set to 0 before proceeding to the next step.
- Now, under the Filter tab in Peptide Settings delimit the length of accepted peptides from 8 to 25 amino acids.
- In Transition Settings, under Filter tab set Precursor Charges as 2,3, set Ion Charges as 1 and set Ion Types to y. Product ions can be selected depending on the user's choice. Select N-terminal to proline for special ions and leave all other parameters as default.
NOTE: The peptide and transition settings can vary according to the experiment. - Insert the peptides or proteins of interest by clicking on Edit and moving to Insert. To insert proteins, copy their accession IDs and to insert specific peptides, copy the peptide sequences.The software maps the accession IDs to the background proteome and the transition list is created based on the peptide and transition settings.
- Export the transition list. Ensure that in the dropdown for Instrument Type, the right instrument is selected. For the optimization experiments, one may choose to split the transition lists into smaller numbers by setting the desired number of transitions per file in Skyline. This will ensure that the instrument is not overwhelmed to screen too many transitions in a single run. The number of transitions needs to be further optimized to get a single method – this we mention henceforth under the method refinement section.
6 LC parameters
- Use a binary solvent system with the aqueous solvent containing 0.1% FA (Solvent A) and the organic solvent containing 80% ACN (Solvent B).
- Set the column temperature to 45 °C.
- Set an LC gradient of 10 min with a 450 µL/min flowrate (as shown in Table S1).
7 MS parameters
NOTE: The explained assay has been developed and optimized for TSQ Altis Triple Quadrupole Mass Spectrometer.
- Optimize runs parameters like Q1 and Q3 resolution, dwell time and cycle time – one parameter at a time. We find that 0.7 resolution for Q1 and Q3 works best. The cycle time or dwell time might need to be tweaked according to the number of transitions and average peak width of the peptides being monitored.
- Use the MS parameters in Table S2 and Table S3. The total method duration is 10 min.
NOTE: The parameters remain same for all the runs during and after method refinement, unless mentioned otherwise. For a fresh experiment, there may be a need to change certain parameters depending on the type of the sample and sample processing steps.
8 Run sequence and Instrument QC
- Prepare a mixture of water: methanol: isopropanol: acetonitrile in 1:1:1:1 ratio. Use this mixture as a blank.
- Prepare peptides for any standard sample that can be used to consistently monitor the performance of the instrument. This will be used as a QC standard. We detect peptides from BSA digests that have been optimized and give good and consistent response over several days (Figure 2).
- To run samples, the sequence should start with a couple of blanks, followed by the QC standard and clinical samples. Always ensure there is one blank between two consecutive samples.
- For the ease of comparison, ensure equal amounts and volumes of each sample are injected every time.
9 Method refinement
- Analyze the preliminary data obtained from the pooled samples using Skyline. Look for the right peak, transitions, and parameters such as peak shape and intensity to select the best results. Save the file as a new Skyline project.
- Use a library to find the best matching peak and consequently the best possible transition list is advised. A library is a set of MS/MS peaks available from literature or in-house experiments.
- Export a fresh transition list from the newly saved Skyline project and use this transition list to make a fresh method. Acquire data from the new method created and repeat the process of refining the transitions using Skyline.
- Once the list of peptides and transitions is finalized following data analysis using Skyline, fine tune the method by trying out different permutations of cycle time and dwell time.
NOTE: The use of heavy labelled synthetic peptides and indexed retention time (iRT) peptides makes method refinement easy8, 9. It is hence advisable that fine tuning of the method be performed using these peptides. - Using the retention time information from the refinement experiments, create a final method that is scheduled (having a defined acquisition window).
- Prepare samples for individual subjects. Data will be acquired for these samples using the final scheduled method.
- Once the data is acquired, further downstream analysis and group-wise comparison can be performed by importing the raw files to Skyline.
Representative Results
We performed relative quantification of 3 proteins from 10 samples, 5 samples from each group of patients with abnormalities in the brain. These proteins included Apolipoprotein A-I (APOA-I), Vimentin (VIM) and Nicotinamide phosphoribosyltransferase (NAMPT) which are known to perform diverse roles in the brain cells. Post-run analysis of the data was performed using Skyline-daily (Ver 20.2.1.286). A total of 10 peptides corresponding to 3 proteins were monitored. These included 3 peptides for APOA-I, 4 peptides for VIM and 3 peptides for NAMPT. The total number of transitions from these 10 peptides amounted to 57. The samples were grouped into either of the two groups depending on the condition they belonged to. Using the group comparisons feature of skyline, the peak abundances of these peptides were compared, and relative quantification values were calculated (Figure 3).
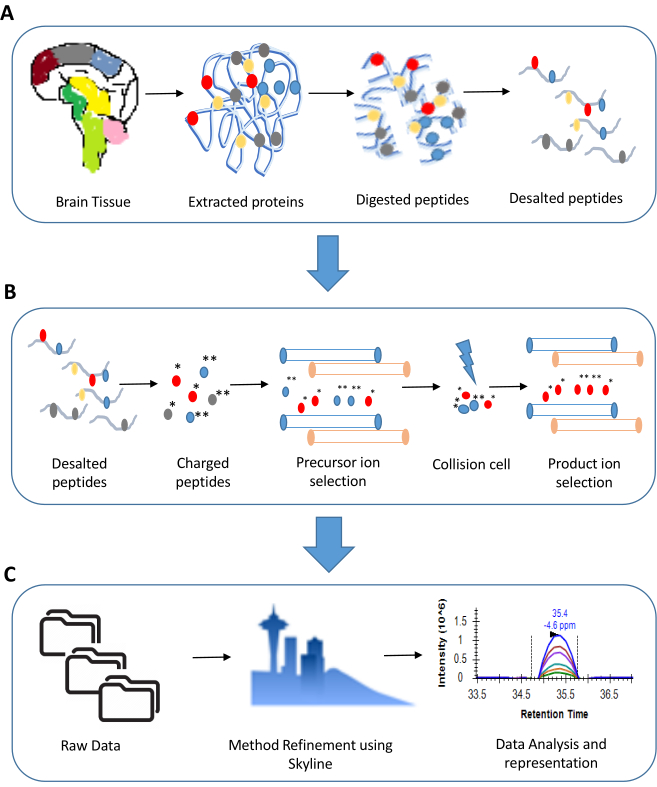
Figure 1: An overview of steps involved in a Multiple Reaction Monitoring experiment. A. Sample preparation for a typical proteomics experiment involves extraction of proteins (for illustration we have shown tissue sample) followed by digestion using trypsin. The digested peptides are ultimately desalted and made LC-MS ready. B. The steps involved in an MRM experiment include precursor and product ion selection based on their m/z values. Only the transitions showing good response are considered for analysis. C. The data analysis in an MRM experiment includes a detailed examination of peak shapes and peak areas. This is ultimately followed by statistical analysis of the results. Please click here to view a larger version of this figure.
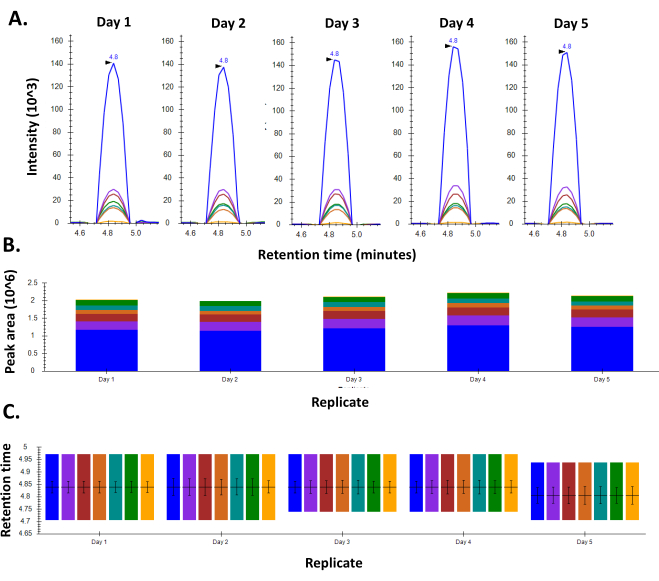
Figure 2: Consistency in response for BSA using an optimized MRM method. A. Chromatogram for a representative peptide of BSA shows consistent peak shape and intensity throughout the five days the experiment was performed. B. Retention time consistency observed for the peptide on all the five days of the experiment C. Peak areas for the peptide as seen over the course of five days in the week. Please click here to view a larger version of this figure.
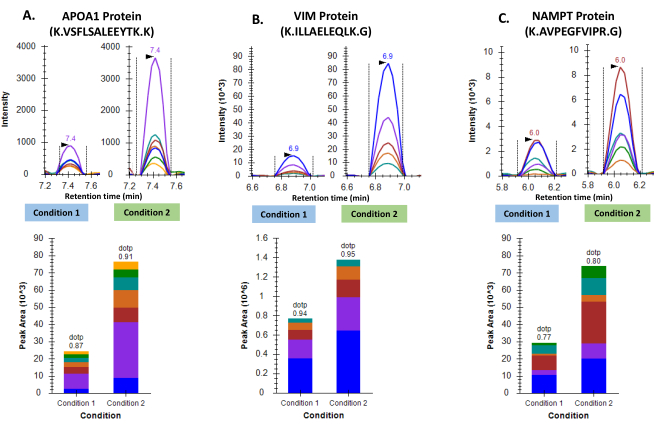
Figure 3: Differential regulation of three proteins in two groups of GBM tumor samples. A. Representative chromatograms for Apolipoprotein A-I and cumulative peak area as seen following inter-group comparison. B. Representative chromatograms for Vimentin and cumulative peak area as seen following inter-group comparison. C. Representative chromatograms for Nicotinamide phosphoribosyltransferase and cumulative peak area as seen following inter-group comparison. Please click here to view a larger version of this figure.
Table S1: Details of 10-minute LC gradient to be used for all samples. Please click here to download this Table.
Table S2: Parameter settings for the ion source. Please click here to download this Table.
Table S3: Parameter settings for MRM method. Please click here to download this Table.
Discussion
Techniques like Immunohistochemistry and Western blotting were considered as the gold standards for validation of protein targets for many years. These methods find use even today with minor modifications in the protocol and little dependence on technology making them very cumbersome and tedious. Besides this, they also involve the use of expensive antibodies which do not always show the same specificity across batches and require a great deal of expertise. Additionally, only a small fraction of proteins identified using high throughput techniques like mass spectrometry, have compatible antibodies available, further complicating the whole procedure. Hence targeted proteomic assays are slowly being taken up as the new approach for validating targets10. With most of the target discovery happening on high-throughput omics platforms, panels of validated targets are also being considered for clinical screening applications11,12,13.
The representative results in this article have validated the differential expression of proteins Apolipoprotein A-I (APOA-I), Vimentin (VIM) and Nicotinamide phosphoribosyltransferase (NAMPT) in two conditions (condition 1 and condition 2) of the brain tissue. Apolipoprotein A-I has been reported to play a pivotal role in maintenance of cerebrovascular integrity and reducing the risk of Alzhiemer's disease. Even though ApoA-I is not synthesized in the brain, its ability to cross the blood brain barrier (BBB) makes its presence in the brain vital14. The Vimentin protein has been studied in a number of roles inside the brain. However, one of the key functions of Vimentin is its involvement in microglia activation. Reduced expression of Vimentin was associated with impairment of microglial activation15. The protein NAMPT has been reported to play a key role in ageing related loss of neurons and cerebral vascular endothelial dysfunctions16. All the three proteins have been reported to play a multitude of roles in normal brain cells and brain related malignancies. Therefore, MRM based validation for these proteins and their peptides can find great use in clinical diagnosis related to various brain related disorders.
A fully optimized targeted assay can be easily used for high-throughput detection and quantification of a target panel. The rate limiting step is the initial method optimization which is tedious and varies based on the sample type, protein/peptide targets, instrument being used and the detection bias of certain peptides. It is crucial that the transition list is optimized for a robust assay. Any user interested in developing such an assay for human brain tissue samples will find that the above explained protocol minimizes these variable factors. It describes an optimized protocol for peptide extraction from this unique and tedious tissue biospecimen and optimal parameters to be used in the instrument with special attention to crucial quality control steps at various points of the protocol. As with any new technology, the researchers have provided a set of guidelines, which authors need to furnish or what steps they follow during the experiment. To this front, in 2017, the MCP guidelines for reporting targeted proteomics assays and data were laid down17. These guidelines ensure that the reported study/assay is reliable and reproducible, hence increasing the applicability of the method. By taking the right precautions and utilising the true potential of this assay, researchers would soon be able to come up with clinically relevant assays with immense potential in diagnosis and therapeutics.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We acknowledge MHRD-UAY Project (UCHHATAR AVISHKAR YOJANA), project #34_IITB to SS and MASSFIITB Facility at IIT Bombay supported by the Department of Biotechnology (BT/PR13114/INF/22/206/2015) to carry out all MS-related experiments.
We extend our special thanks to Mr. Rishabh Yadav for making and editing of the entire video and Mr. Nishant Nerurkar for his work in editing the audio.
Materials
| Reagents | |||
| Acetonitrile (MS grade) | Fisher Scientific | A/0620/21 | |
| Bovine Serum Albumin | HiMedia | TC194-25G | |
| Calcium chloride | Fischer Scienific | BP510-500 | |
| Formic acid (MS grade) | Fisher Scientific | 147930250 | |
| Iodoacetamide | Sigma | 1149-25G | |
| Isopropanol (MS grade) | Fisher Scientific | Q13827 | |
| Magnesium Chloride | Fischer Scienific | BP214-500 | |
| Methanol (MS grade) | Fisher Scientific | A456-4 | |
| MS grade water | Pierce | 51140 | |
| Phosphate Buffer Saline | HiMedia | TL1006-500ML | |
| Protease inhibitor cocktail | Roche Diagnostics | 11873580001 | |
| Sodium Chloride | Merck | DF6D661300 | |
| TCEP | Sigma | 646547 | |
| Tris Base | Merck | 648310 | |
| Trypsin (MS grade) | Pierce | 90058 | |
| Urea | Merck | MB1D691237 | |
| Supplies | |||
| Hypersil Gold C18 column | Thermo | 25002-102130 | |
| Micropipettes | Gilson | F167380 | |
| Stage tips | MilliPore | ZTC18M008 | |
| Zirconia/Silica beads | BioSpec products | 11079110z | |
| Equipment | |||
| Bead beater (Homogeniser) | Bertin Minilys | P000673-MLYS0-A | |
| Microplate reader (spectrophotometer) | Thermo | MultiSkan Go | |
| pH meter | Eutech | CyberScan pH 510 | |
| Probe Sonicator | Sonics Materials, Inc | VCX 130 | |
| Shaking Drybath | Thermo | 88880028 | |
| TSQ Altis mass spectrometer | Thermo | TSQ02-10002 | |
| uHPLC – Vanquish | Thermo | VQF01-20001 | |
| Vacuum concentrator | Thermo | Savant ISS 110 |
References
- Picotti, P., Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nature Methods. , (2012).
- Carr, S. A., et al. Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Molecular and Cellular Proteomics. 13 (3), 907-917 (2014).
- Pitt, J. J. Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. The Clinical biochemist Reviews. 30 (1), 19-34 (2009).
- Hosp, F., Mann, M. A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron. 96 (3), 558-571 (2017).
- Scopes, R. K. Measurement of protein by spectrophotometry at 205 nm. Analytical Biochemistry. , (1974).
- Kusebauch, U., et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell. , (2016).
- MacLean, B., et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26 (7), 966-968 (2010).
- Gerber, S. A., Rush, J., Stemman, O., Kirschner, M. W., Gygi, S. P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proceedings of the National Academy of Sciences of the United States of America. , (2003).
- Escher, C., et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. , (2012).
- Gillette, M. A., Carr, S. A. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nature Methods. 10 (1), 28-34 (2013).
- Whiteaker, J. R., et al. A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nature Biotechnology. , (2011).
- Hüttenhain, R., et al. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Science Translational Medicine. 4 (142), 94 (2012).
- Mermelekas, G., Vlahou, A., Zoidakis, J. SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine. Expert Review of Molecular Diagnostics. 15 (11), 1441-1454 (2015).
- Koldamova, R. P., Lefterov, I. M., Lefterova, M. I., Lazo, J. S. Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits Aβ aggregation and toxicity. Biochemistry. , (2001).
- Jiang, S. X., Slinn, J., Aylsworth, A., Hou, S. T. Vimentin participates in microglia activation and neurotoxicity in cerebral ischemia. Journal of Neurochemistry. , (2012).
- Liu, L. Y., et al. Nicotinamide Phosphoribosyltransferase May Be Involved in Age-Related Brain Diseases. PLoS ONE. , (2012).
- Abbatiello, S., et al. New guidelines for publication of manuscripts describing development and application of targeted mass spectrometry measurements of peptides and proteins. Molecular and Cellular Proteomics. 16 (3), 327-328 (2017).
