La cristallographie macromoléculaire à température ambiante (RT) est à nouveau populaire au sein de la communauté de la biologie structurale. Le développement de sources lumineuses à rayons X (XFEL) a stimulé le développement d’approches de livraison d’échantillons RT 1,2,3,4, et ces méthodes ont maintenant été appliquées aux synchrotrons 5,6,7,8. Non seulement les méthodes RT ouvrent la possibilité de stratégies expérimentales pompe-sonde 9,10,11,12, mais il y a aussi de plus en plus de preuves qu’elles favorisent des états conformationnels alternatifs dans les protéines 13,14,15,16,17.
Cependant, la principale raison pour laquelle les méthodes cryogéniques ont gagné du terrain par rapport aux approches RT à la fin des années 1990 était le ralentissement des dommages causés par les rayonnements par des températures cristallines inférieures à zéro18. Les méthodes cryogéniques19 ont commencé à permettre la collecte d’un ensemble de données complet à partir d’un seul cristal de protéine. Les méthodes modernes de RT dans les XFEL et les synchrotrons ont résolu le problème des dommages causés par le rayonnement monocristallin par le développement de stratégies d’administration rapide (> 100 Hz)de cristaux 1,2,3,4. Ces méthodes permettent de collecter un ensemble de données complet à partir de milliers de cristaux exposés individuellement. Ces approches d’administration RT nécessitent donc la production de grandes quantités de solutions contenant des microcristaux homogènes (> 100 μL de cristaux < 50 μm). Cependant, étant donné que les cryo-méthodes ont tendance à ne nécessiter que des monocristaux, les méthodes de création de telles boues microcristallines ne sont actuellement pas omniprésentes dans les laboratoires de cristallographie des protéines.
Il existe des exemples dans la littérature sur la façon de faire des parties de la procédure d’optimisation de la microcristallisation pour les échantillons de cristallographie en série. Ici, une distinction doit être faite entre les protéines membranaires et solubles. Les protocoles visant à optimiser la croissance des cristaux de protéines micro-membranaires cultivés dans la monooléine (ou un autre lipide), pour la phase cubique lipidique (LCP), ont été bien décrits20,21,22. Cependant, les méthodes de microcristallisation des protéines solubles, y compris les protéines membranaires cultivées dans des conditions non LCP, font généralement défaut. Des études antérieures se sont concentrées sur des parties spécifiques du processus, telles que le criblage de microcristaux 23,24, l’amélioration de la nucléation 24 et la mise à l’échelle à l’aide de la diffusion à interface libre 25, mais pas une méthode complète.
Cependant, une méthode a récemment été décrite26 qui tente d’offrir un protocole complet. Comme beaucoup d’aspects de la cristallographie des protéines, elle n’est pas nouvelle. Bon nombre des idées proposées ont déjà été décrites par Rayment (2002)27. La méthode vise à montrer aux cristallographes comment effectuer la conversion d’un seul cristal cultivé par diffusion de vapeur en une méthodologie de lot pour cultiver des milliers de microcristaux. La méthode se concentre sur la diffusion de vapeur comme point de départ commun, car 95% de tous les dépôts de banques de données sur les protéines (PDB) proviennent de cristaux cultivés dans des plaques de diffusion de vapeur26. La diffusion de vapeur n’est cependant pas la méthode idéale pour la micro-cristallisation26, c’est pourquoi une méthodologie est décrite pour convertir la diffusion de vapeur en cristallisation par lots. Une fois que les cristaux peuvent être cultivés par lots, les voies de mise à l’échelle vers de plus grands volumes deviennent plus pratiques. Compte tenu des aléas de la cristallisation des protéines, les auteurs soulignent que cette méthode n’est pas infaillible. Cependant, le protocole devrait, au moins, fournir un aperçu de « l’espace de cristallisation » d’une protéine.
Cette méthode repose sur le diagramme de phase de cristallisation des protéines et sur la façon dont la compréhension de ce diagramme peut servir de guide lors de l’optimisation de la micro-cristallisation. Un diagramme de phase protéique est généralement représenté comme un diagramme x/y avec des concentrations de précipitants et de protéines sur les axes x et y, respectivement (Figure 1A). À partir du point d’eau pure (coin inférieur gauche – Figure 1A), la concentration de protéines et de précipitants augmente jusqu’à ce que la ligne de solubilité soit atteinte. La ligne de solubilité marque le point de sursaturation (ligne violette – Figure 1A). Lorsqu’une protéine est sursaturée, la solution devient thermodynamiquement instable et commence à se séparer en deux phases: « riche en protéines » et une solution saturée stable. Cette séparation peut se produire n’importe où au-delà de la ligne de solubilité et sa cinétique dépend des propriétés de la protéine et des composants de la solution.
Lorsque les concentrations de protéines et de précipitants sont trop élevées, la protéine se décompose de manière instable hors de la solution et donne un précipité amorphe (région rose – Figure 1A). Cependant, une séparation de phase ordonnée peut se produire dans la région de nucléation [voir Garcia-Ruiz (2003)28 pour une description détaillée] et les nucléants cristallins ont la propension à se former (région verte – Figure 1A). La nucléation et la croissance éliminent les protéines de la solution et déplacent la goutte dans la région métastable où la croissance peut se poursuivre jusqu’à ce que la ligne de solubilité soit atteinte [voir McPherson et Kuznetsov (2014)29 pour une discussion détaillée]. Le diagramme est, pour la grande majorité des conditions de cristallisation, une simplification grossière30. Indépendamment de cela, le diagramme est toujours d’une grande utilité pour les micro-cristallographes car la cartographie du diagramme permet de déterminer la ligne de solubilité et la cinétique de nucléation.
En termes de création de micro-cristaux, les deux facteurs lors de la cristallisation qui doivent être optimisés sont le nombre de cristaux (Xn) et leur dimension moyenne la plus longue (Xs). X n sera proportionnel au nombre d’événements de nucléation (n ) (Eq. 1).
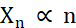 Éq. 1
Éq. 1
X s est proportionnelle à la concentration de protéines libres au-dessus de la ligne de solubilité (Ps) divisée par Xn (Eq. 2).
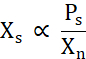 Éq. 2
Éq. 2
Dans une situation parfaite, chaque événement de nucléation produirait un cristal possible et chacun de ces cristaux aurait un accès égal à la protéine disponible en solution. La figure 2 est une représentation graphique d’un scénario idéal de la relation entre Xn et Xs. En pratique, le principal contrôle qu’un cristallographe a sur Xn et X s est en influençant la quantité de nucléation ou par l’ajout de cristaux graines. Le microcristallographe doit juger comment augmenter Xn de telle sorte qu’une concentration cristalline et une taille cristalline appropriées puissent être créées.
La majorité des techniques de cristallisation nécessitent une « période de transition » (Figure 1B). Par exemple, dans une expérience de diffusion de vapeur, lors du mélange de la protéine et des solutions précipitantes, les concentrations de chacune changeront à mesure que la goutte s’équilibre avec la solution du puits. On espère que ces changements feront progressivement passer la goutte dans la zone de nucléation où la propension à la cristallisation augmentera. Au fur et à mesure que les cristaux commencent à nucléer et à croître, la quantité de protéines en solution commence à diminuer, ce qui diminue la probabilité d’une nucléation supplémentaire. La quantité ultime de nucléation sera spécifique à la protéine et à la condition, et dépendra également de la profondeur de pénétration dans la zone de nucléation. Compte tenu de la pénétration limitée de la zone de nucléation des méthodes qui nécessitent une étape de transition, le niveau de nucléation sera finalement limité au taux de nucléation à la limite de la région de nucléation métastable.
En raison de l’importance de pouvoir améliorer le niveau de nucléation pour un micro-cristallographe, il est important de passer à une méthodologie de cristallisation par lots. Le lot peut tirer davantage parti de l’ensemble de la région de nucléation (Figure 1C). Dans les méthodes discontinues, l’idée est de mélanger la protéine et le précipitant ensemble de sorte qu’une solution sursaturée soit créée sans qu’il soit nécessaire de modifier les concentrations des composants. La nucléation devrait être possible immédiatement après le mélange. Les méthodes batch permettent donc d’atteindre théoriquement toute la zone de nucléation. Toute augmentation de la cinétique de nucléation au-delà de la limite de nucléation métastable peut alors être utilisée.
Si le niveau basal de nucléation cristalline n’est pas suffisant pour générer un grand Xn, des méthodes de micro-ensemencement peuvent être utilisées. Dans le micro-ensemencement, les cristaux pré-cultivés sont brisés pour créer une boue de fragments cristallins qui peut agir comme un échafaudage pour la croissance des cristaux frais31,32. Le micro-ensemencement a été largement utilisé dans la préparation d’échantillons cristallographiques en série comme moyen d’augmenter Xn sans avoir besoin d’augmenter la nucléation cristalline (Figure 1C).
La transition de la diffusion de vapeur au lot peut être visualisée sur un diagramme de phase comme déplaçant le point de départ expérimental des régions non sursaturées ou métastables vers la zone de nucléation. Cela peut être fait en augmentant les concentrations de protéines et/ou de précipitants, et/ou le rapport des deux dans la goutte (Figure 1D), et en observant quelles conditions produisent des cristaux apparaissant rapidement (< 24 h)26. L’équilibrage complet des gouttes de diffusion de vapeur peut prendre des jours ou des semaines33. Par conséquent, en recherchant des conditions qui montrent des cristaux apparaissant rapidement, les conditions des lots peuvent être trouvées sans avoir à passer à d’autres formats de criblage de cristallisation tels que le micro-lot34,35,36,37.
Une fois que la zone de nucléation a été trouvée, une condition de lot a été trouvée et un morphogramme – ici, un diagramme de phase approximatif – peut être créé. Le morphogramme est d’une grande utilité lorsqu’il s’agit d’utiliser un protocole de lot ensemencé ou de lot direct. En traçant le Xn en fonction de la concentration en protéines et en précipitants, on peut évaluer la cinétique de nucléation26. Si X n reste faible dans toute la région de nucléation, un lot ensemencé peut être nécessaire pour rendre Xn assez grand pour limiter la croissance cristalline. Cette évaluation est la première étape du processus de mise à l’échelle vers des volumes plus importants (> 100 μL).
Cette méthode a été conçue de manière à pouvoir être réalisée dans la majorité des laboratoires de cristallisation en utilisant un équipement standard de cristallisation par diffusion de vapeur. De nombreuses études ont également été menées qui décrivent des techniques pour faciliter de nombreuses parties de ce processus, si l’équipement est disponible. Ceux-ci incluent, mais ne sont pas limités à, la diffusion dynamique de la lumière (DLS)25,27, l’imagerie non linéaire 20,24,25, la diffraction des poudres 20,24,27 et la microscopie électronique 26 [voir Cheng et al. (2020)40 pour une belle revue].
L’objectif de ce travail est de fournir une démonstration visuelle de la méthode de transition de la cristallisation par diffusion de vapeur en petit volume ( 100 μL). L’endothiapepsine de Cryphonectria parasitica a été utilisée comme exemple de système pour démontrer cette traduction. Le type d’expérience et la méthode d’administration d’échantillons pour lesquels les microcristaux sont nécessaires influenceront la sortieidéale de Xs 26. Pour les expériences de mélange nécessitant une résolution temporelle de l’ordre de la milliseconde41 ou des buses virtuellesdynamiques en phase gazeuse42, un X s final de < 5 μm peut être souhaitable. Dans ce cas, l’objectif était de produire des cristaux de protéines diffractés à environ 1,5 Å, pour une expérience pompe-sonde activée par photon, et en utilisant une approche de livraison à cible fixe.
Pour illustrer les besoins en échantillons d’une telle expérience de cristallographie en série utilisant l’endothiapepsine, le tableau 1 montre les paramètres expérimentaux d’une expérience hypothétique. L’échantillon d’information était basé sur le protocole décrit ci-dessous. Compte tenu de certaines estimations prudentes sur les taux de réussite et les exigences de collecte de données, 50 mg est l’estimation de la consommation totale de l’échantillon pour l’ensemble de l’expérience.
La figure 3 montre un organigramme du processus d’optimisation complet depuis la cristallisation initiale par diffusion de vapeur à petit volume jusqu’au lot à grande échelle. Pour la majorité des projets de cristallographie en série, ce protocole commencera à l’étape 2 : « transition vers le lot », puisque la protéine cible aura déjà été cristallisée. Cependant, l’étape 1 a été incluse pour être complète et pour rappeler aux lecteurs son importance. Trouver une condition qui donne lieu à un cristal simple, grand et bien diffractant est le meilleur point de départ pour l’optimisation des microcristaux. À l’étape 2, cette condition peut ensuite être optimisée de la diffusion de vapeur au lot, et un morphogramme des régions de nucléation et métastables peut être tracé. Une fois cette opération effectuée, la mise à l’échelle de la condition de lot à des volumes plus importants peut être effectuée à l’étape 3. À la fin de l’organigramme, un cristallographe aura créé un protocole de microcristallisation reproductible et de grand volume (> 100 μL) pour l’endothiapepsine. Cette méthode peut ensuite être appliquée à leur protéine d’intérêt particulière.
