Cell Migration and Cell Adhesion Assays to Investigate Leishmania-Host Cell Interaction
Summary
Here we study implications of Leishmania-host interaction by exploring Leishmania-infected dendritic cells migration. The differentiation and infection of dendritic cells, migration analysis, and the evaluation of adhesion complexes and actin dynamics are described. This method can be applied to other host cell migration studies when infected with Leishmania or other intracellular parasite species.
Abstract
Leishmania is an intracellular protozoan parasite that causes a broad spectrum of clinical manifestations, ranging from self-resolving localized cutaneous lesions to a highly fatal visceral form of the disease. An estimated 12 million people worldwide are currently infected, and another 350 million face risk of infection. It is known that host cells infected by Leishmania parasites, such as macrophages or dendritic cells, can migrate to different host tissues, yet how migration contributes to parasite dissemination and homing remains poorly understood. Therefore, assessing these parasites’ ability to modulate host cell response, adhesion, and migration will shed light on mechanisms involved in disease dissemination and visceralization. Cellular migration is a complex process in which cells undergo polarization and protrusion, allowing them to migrate. This process, regulated by actin and tubulin-based microtubule dynamics, involves different factors, including the modulation of cellular adhesion to the substrate. Cellular adhesion and migration processes have been investigated using several models. Here, we describe a method to characterize the migratory aspects of host cells during Leishmania infection. This detailed protocol presents the differentiation and infection of dendritic cells, the analysis of host cell motility and migration, and the formation of adhesion complexes and actin dynamics. This in vitro protocol aims to further elucidate mechanisms involved in Leishmania dissemination within vertebrate host tissues and can also be modified and applied to other cell migration studies.
Introduction
Leishmaniasis, a neglected tropical disease caused by protozoan parasites belonging to the genus Leishmania, results in a wide-ranging spectrum of clinical manifestations, from self-healing localized cutaneous lesions to fatal visceral forms of the disease. It has been estimated that up to one million new leishmaniasis cases arise annually, with a reported 12 million people currently infected worldwide1. Visceral leishmaniasis (VL), which can be fatal in over 95% of cases when left untreated, causes more than 50,000 deaths annually, affecting millions in South America, East Africa, South Asia, and the Mediterranean region2. The main etiological agent of VL in the new world, Leishmania infantum, is transmitted to humans by infected female sandflies during blood-feeding3. These parasites are recognized and internalized by phagocytes, e.g., macrophages and dendritic cells3,4,5. Inside these cells, parasites differentiate into their intracellular forms, known as amastigotes, which will then multiply and be transported via the lymphatic system and bloodstream to different host tissues6,7. However, the mechanisms by which Leishmania parasites are disseminated in the vertebrate host, as well as the role played by host cell migration in this process, remain unclear.
Cell migration is a complex process executed by all nucleated cells, including leukocytes8. According to the classic cycling model of cell migration, this process involves several integrated molecular events that can be divided into five steps: leading-edge protrusion; adhesion of the leading edge to matrix contacts; contraction of cellular cytoplasm; release of the rear edge of the cell from contact sites; and the recycling of membrane receptors from the rear to the front of the cell9.
For cell migration to occur, protrusions must be formed and then stabilized through attachment to the extracellular matrix. Among the different receptor types involved in the promotion of cell migration, integrins are notable. Integrin activation results in migration-related signaling; intracellular signaling then occurs via focal adhesion kinase (FAK) and Src family kinases, in addition to talin, vinculin, and paxillin molecues10,11,12. The phosphorylation of paxillin by activated kinases, including FAK, leads to the recruitment of effector molecules, which transduces external signals that prompt cell migration. It has been shown that paxillin is an intracellular molecule that is crucial to cell adhesion, actin polymerization, and cell migration processes13,14,15.
The actin cytoskeleton plays a central role in the polarization and migration of phagocytes16. During cell migration process, protrusions formed due to actin polymerization become stabilized through cell adhesion to the extracellular matrix. This process may be modulated by integrin receptors associated with the actin cytoskeleton17,18,19. Several actin-binding proteins regulate the rate and organization of actin polymerization in cellular protrusions20. Studies have shown that RhoA, Rac, and Cdc42 regulate actin reorganization after the stimulation of adherent cells by extracellular factors21,22. During migration, Rac1 and Cdc42 are located at the leading edge of the cell, controlling the extension of lamellipodia and filopodia, respectively, while RhoA, located at the rear of the cell, regulates the contraction of the actomyosin cytoskeleton15,23,24,25.
Studies have shown that Leishmania infection modulates host cell functions, such as adhesion to the cellular substrate and migration26,27,28,29,30,31. Immature DCs reside in peripheral tissues; upon interaction with PAMPS, these cells become activated and migrate to the draining lymph nodes, prompting antigen presentation to T cells. A previous study using a mouse model showed that L. amazonensis infection provokes a reduction in the migration of DCs to draining lymph nodes29. It was also demonstrated that the inhibition of the adhesion process reduced DC migration after infection with L. major30. Nonetheless, the impact of DC migration on parasite dissemination in the host, as well as the mechanisms involved in this process, remain poorly understood.
Here we present a compiled step-by-step protocol to perform an in vitro adhesion and migration assay involving human DCs infected by Leishmania. This method comprises not only the differentiation and infection of DCs, but also permits the analysis of host cell motility and migration, the formation of adhesion complexes, as well as actin dynamics. The presently described in vitro protocol allows researchers to further investigate the mechanisms involved in Leishmania dissemination within vertebrate host tissues and can also be manipulated and applied to other cell migration studies.
Protocol
The procedures described herein were approved by the Institutional Review Board of the Gonçalo Moniz Institute (IGM-FIOCRUZ, protocol no. 2.751.345). Blood samples were obtained from healthy volunteer donors. Animal experimental procedures were conducted in accordance with the Ethical Principles in Animal Research adopted by the Brazilian law 11.784/2008 and were approved and licensed by the Ethical Committee for Animal Research of the Gonçalo Moniz Institute (IGM-FIOCRUZ, protocol no. 014/2019).
1. Isolation and differentiation of human dendritic cells
- Pipette 10 mL of the density gradient medium in 50 mL centrifuge tubes.
- Label the 50 mL tubes according to each donor's sample.
- Collect ~ 50 mL of venous blood from healthy donors and after collection, follow the procedures described below in the flow cabinet.
- Carefully transfer the collected blood into centrifuge tubes and dilute in saline solution (0.9% sodium chloride) in a ratio of 1:1 at room temperature.
- Slowly overlay the diluted blood on the top of the density gradient medium.
- Centrifuge tubes containing the blood and density gradient medium at 400 x g for 30 min at room temperature.
NOTE: Switch off the brake before centrifuging to prevent the mixing of gradient layers. After the 1st centrifugation, lower the centrifuge temperature to 4 °C. - Carefully remove the tubes from the centrifuge.
- Identify the ring of peripheral blood mononuclear cells (PBMC) in the sample (buffy coat); gently remove the residual plasma with a pipette.
NOTE: After centrifugation, the following gradient layers will have formed: erythrocytes, density gradient medium, PBMC ring, and plasma. The PBMC ring is located between the density gradient medium and plasma layers. - Transfer the cloud-like PBMC layer to a new tube and bring up the volume to 30 mL with saline solution.
- Centrifuge the tubes containing cell suspension at 250 x g for 10 min at 4 °C. Discard the supernatant and resuspend the pellet in 1 mL of saline solution.
- Collect an aliquot for cell counting with the trypan blue exclusion method. First, dilute at a ratio of 1:1,000. Then, use 10 µL of the diluted cells for trypan blue staining and count cells using a Neubauer chamber to determine cell viability.
- Centrifuge again at 200 x g for 10 min under 4 °C.
- Resuspend the pellet in MACS buffer. Use 80 µL of the buffer for every 1 x 107 cells.
NOTE: MACS buffer composition: Prepare a solution containing phosphate-buffered saline (PBS), pH 7.2, 0.5% bovine serum albumin (BSA), and 2 mM EDTA. Dilute the buffer solution at a 1:20 ratio with the rinsing solution. Keep the buffer cold and store at 2-8 °C. - Add CD14 microbeads to the cell suspension prepared in step 1.13. Use 20 µL of CD14 microbeads for every 1 x 107 cells.
NOTE: CD14 microbeads are used for the positive selection of human monocytes from PBMCs, as beads bind to CD14-positive cells expressed on most monocytes. - Homogenize the solution containing the pellet and microbeads using a pipette. Keep on ice for 15 min.
- Centrifuge the suspension at 300 x g for 10 min under 4 °C.
- Resuspend cells in MACS buffer. Use 1-2 mL for 1 x 107 cells in the cell microbead mixture.
- Centrifuge the suspension at 300 x g for 10 min under 4 °C.
- Aspirate the supernatant and resuspend the pellet in 500 µL of MACS buffer.
NOTE: This is the maximum volume of the cell suspension that can be processed through one column. - Assemble the magnetic column.
- Wash the column once by adding 500 µL of MACS buffer. Allow buffer to flow under gravity through the column.
NOTE: Do not allow the column to dry, as any air that enters can obstruct the column. - Add 500 µL of the cell sample solution from step 1.19 per column. Allow the cell sample solution to flow under gravity through the column.
- Wash the column by adding 500 µL of MACS buffer (2x). Only add the fresh buffer when the column reservoir is empty. Avoid drying.
- Pipette 1 mL of the MACS bufferonto the column and place it in a new tube underneath. Immediately flush out the magnetically labeled cells by firmly pushing the plunger into the column.
- Centrifuge the CD14 enriched cells at 300 x g for 10 min under 4 °C.
- Count cells using a Neubauer chamber.
- Resuspend cells in 1 mL complete RPMI with interleukin-4 (IL-4) (100 µL/mL) + granulocyte-macrophage colony-stimulating factor (GM-CSF) (50 ng/mL).
NOTE: GM-CSF is a cytokine secreted by macrophages, T cells, mast cells, natural killer cells, endothelial cells, and fibroblasts that induces the differentiation and proliferation of myeloid progenitors in the bone marrow. GM-CSF and IL-4 are used to induce dendritic cell differentiation32. - Seed cells in a 24 well plate at a concentration of 2 × 105 cells per well in 500 μL of complete RPMI medium containing and incubate the cells for 7 days in the cell culture incubator at 37 °C.
2. Leishmania infantum cultivation
NOTE: Leishmania infantum (MCAN/BR/89/BA262) parasites are used in this assay. Hamsters were intravenously infected with 20 µL of the solution containing 1 x 106 L. infantum promastigotes in sterile saline. After 1 to 2 months, animals were euthanized and the amastigote forms of Leishmania were recovered from their spleens and differentiated into promastigotes33.
- Grow L. infantum promastigotes isolated from previously infected hamster spleens in an inclined 25 cm2 cell culture flask containing 3 mL of Novy-Nicolle-MacNeal medium (NNN) and 5 mL of supplemented Minimum Essential Medium (MEM).
NOTE: In this assay, MEM medium was supplemented with 10% fetal bovine serum (FBS) and 24.5 nM hemin bovine. - Incubate at 24 °C for 7 days in BOD (Bio-Oxygen Demand) incubator.
- Pipette 100 µL of promastigote culture into a new 25 cm2 cell culture flask containing 5 mL of supplemented MEM medium.
- Incubate the promastigote culture at 24 °C in BOD incubator until promastigotes reach the stationary phase. Periodically count cultured promastigotes by transferring an aliquot of parasite suspension diluted in saline to a Neubauer chamber (i.e., hemocytometer) to determine the stationary phase of growth.
NOTE: It is not recommended to use first-passage parasites after cultivation in NNN medium to avoid residual traces of rabbit blood. - Transfer 1 x 105 of stationary growth phase culture of L. infantum promastigotes to a new 25 cm2 cell culture flask and add 5 mL of MEM supplemented medium.
- Monitor the growth of the culture periodically for approximately 7 days using a Neubauer chamber until the stationary phase is achieved. Parasites are now ready for use in experimental infection procedures.
NOTE: Promastigote cultures are considered viable for infection for up to 7 passages in vitro; additional passages may induce loss of virulence.
3. Human dendritic cell infection
- Inside a biological laminar flow cabinet, transfer all the contents from the parasite culture flask to 50 mL centrifuge tubes.
- Add cold saline solution to a final volume of 40 mL.
- Centrifuge at 1,600 xg for 10 min under 4 °C.
- Discard the supernatant following centrifugation and resuspend the pellet in 1 mL of cold saline solution (Repeat steps 3.3-3.4 three times).
- After washing the cells to remove any non-viable parasites, resuspend the pellet in 1 mL of cold saline solution.
- Pass the contents slowly through a 1 mL syringe with a 16 G needle 5 times to separate the parasite clusters.
- Remove an aliquot to determine the parasite concentration in a hemocytometer (calculate the mean number of parasites in 4 quadrants x dilution factor x 104).
- Wash dendritic cells by adding 1 mL of saline solution and centrifuging cells at 300 x g for 10 min at room temperature (3 times).
- Calculate the quantity of L. infantum required for the experimental infection; ratio: 20 parasites per cell.
- Place the required volume of L. infantum in each well of cell culture plates.
- Incubate dendritic cells with parasites for 4 h in an incubator at 37 °C under 5% CO2.
- After step 3.11, centrifuge plates at 300 x g for 10 min under 4 °C.
4. Migration assay using cell culture inserts
- After infection, wash dendritic cells (infected or not) 3 times with 1 mL of saline solution at room temperature to remove non-internalized parasites. After each wash, centrifuge plates at 300 x g for 10 min under 4 °C.
- Once non-internalized parasites are removed, detach cells by adding 200 µL of the cell dissociation reagent in each well and incubate for 15 min. Keep the cells in an incubator at 37 °C under 5% CO2.
- Homogenize cells using a 1,000 µL pipette tip to allow cells to loosen.
- Transfer the dissociated cells to a 50 mL centrifuge tube.
- Centrifuge at 300 x g for 10 min under 4 °C.
- Resuspend the pellet in 1 mL of RPMI medium supplemented with 10% FBS.
- Pass the contents of each tube slowly through a 1 mL syringe with a 25 G needle 5 times to separate cells.
- Remove an aliquot, dilute using trypan blue to determine the cell concentration using a hemocytometer (mean of viable cells in 4 quadrants × dilution factor × 104).
- Preparation of cell culture microplate inserts:
NOTE: Cell culture inserts (5.0 µM pore polycarbonate membrane) are recommended for migration assays using dendritic cells, macrophages, and monocytes, as these inserts allow phagocytes to pass through the membranes.- Remove inserts with sterile tweezers and place in empty wells.
- Add 600 µL of RPMI medium supplemented with 10% FBS to each well; add chemoattractant chemokine (C-C motif) ligand 3 (CCL3) (1 µL for every 1 mL of medium).
NOTE: CCL3 is a chemokine that regulates DC migration34. - Using sterile tweezers, place inserts in wells containing the medium.
- Add 2 x 105 dendritic cells (infected or not) in 100 µL of medium to each insert.
- Incubate for 4 h to allow cells to migrate.
- After 4 h, take the plate out and aspirate the medium. Add 100 µL of 4% paraformaldehyde and incubate for 15 min.
- Remove paraformaldehyde and add 100 µL of saline solution.
NOTE: Plates can be kept at 4 °C for later assembly. - Collect the supernatant from inserts or from wells to count non-migrating cells or those that crossed the membrane, respectively. Incubate with paraformaldehyde 4% for 15 min.
- Concentrate cells using a cytocentrifugation technique35.
- Add 10 µL of the mounting medium with DAPI to the membranes and place coverslips over the wells.
- Assembly of the insert membrane.
NOTE: This step can be performed outside the biosafety cabinets.- Remove insert membranes from wells.
- Scrape the surface of the insert with a swab to remove any cells that have not migrated.
- Using a scalpel, remove the membrane from the insert.
- Place the membrane on a slide, with cells facing up.
- Add 10 µL of mounting medium with DAPI to each membrane and then place overlay coverslips.
- Cover plates with aluminum foil.
- Incubate plates at room temperature for 30 min, then store at -20 °C
- To analyze cell migration, count the number of cells per field (no less than 10 fields) using a fluorescence microscope at a laser excitation wavelength of 405 nm.
5. Adhesion assay and evaluation of actin polymerization by immunofluorescence
NOTE: For this assay, use 24-well plates with coverslips.
- After infecting cells for 4 h, wash each well 3 times with sterile saline solution at room temperature to remove any non-internalized parasites.
- Centrifuge the plate at 300 x g for 10 min under 4°C. Add 500 µL of 4% paraformaldehyde to each well for 15 min.
- Remove paraformaldehyde and add 1 mL of saline solution.
- Prepare solutions as described in Table 1.
| Primary solutions | Chemical compound | Diluent | |
| Ammonium chloride solution | 0,134 g of NH4Cl | 50 ml of PBS 1X | |
| Saponin 15% | 150 mg of saponin | 1 mL de PBS 1X | |
| Albumin from bovine serum (ABS) 10% | 1 g of ABS | 10 mL of PBS 1X | |
| Secondary solutions | Component 1 | Component 2 | Component 3 |
| Saponin 0,15% | 1mL of saponin 15% | 100 mL of PBS 1X | – |
| PBS 1X / ABS 3% / Saponin 0,15% | 13,8 mL of PBS 1X | 6 mL of ABS 10% | 200 µL of Saponin 15% |
| PBS1X / ABS 0,3% / Saponin 0,15%: | 19,2 mL of PBS 1X | 0,6 mL of ABS 10% | 200 µL of Saponin 15% |
| PBS 1X / ABS 1% / Saponin 0,15% | 17,8 mL of PBS 1X | 2 mL of ABS 10% | 200 µL of Saponin 15% |
Table 1: Buffer recipes.
- Immunostaining
NOTE: The following steps must be performed under agitation.- Wash coverslips 3 times with 1x PBS for 5 min.
- Add 500 µL of ammonium chloride solution per well for 10 min.
- Wash coverslips 3 times with 1x PBS for 5 min.
- Permeabilize the membrane with Saponin 0.15% for 15 min.
- Incubate cells with the blocking solution (PBS 1x / ABS 3% / Saponin 0.15%) for 1 h.
- Wash coverslips 3 times with PBS 1x/Saponin 0.15% for 5 min.
- Dilute primary antibodies in PBS 1x /ABS 1% /Saponin 0.15% as: Rabbit anti-FAK (pTyr397): 1:500 dilution, Rabbit anti-paxilin (pTyr118): 1:100 dilution. Mouse Anti-Rac1: 1:100 dilution. Rabbit Anti-Cdc42: 1:200 dilution. Rabbit Anti-RhoA: 1:200 dilution.
- Incubate cells with primary antibody diluted in PBS 1x/ABS 1% /Saponin 0.15% for 1 h.
NOTE: FAK and Paxillin are key molecules involved in the formation of adhesion complexes13,14,15. The Rho GTPase family is responsible for the organization of the actin cytoskeleton. RAC1 and Cdc42, located at the front edge of the cell, control the formation of lamellipodia and filopodia, respectively; RhoA is located at the rear of the cell and is involved in cytoskeleton contraction15, 23,24,25. - Wash coverslips 3 times with 1x PBS/Saponin 0.15% for 5 min.
- Dilute secondary antibody or phalloidin as follows: Anti-rabbit IgG, Alexa Fluor 568: 1:500 dilution; Anti-mouse IgG, Alexa Fluor 488: 1:500 dilution; Anti-mouse IgG, Alexa Fluor 594: 1:500 dilution; Phalloidin Alexa Fluor 488: 1:1200 dilution.
- Incubate cells with secondary antibody or phalloidin diluted in 1x PBS/ABS 0.3% Saponin 0.15% for 45 min.
NOTE: Incubation of the secondary antibody or phalloidin should be performed in the absence of light. Cover plates with aluminum foil to protect from light during the following steps. - Wash coverslips 3 times with 1x PBS for 5 min.
- Remove coverslips from wells.
- In the absence of light, add 10 µL of mounting medium with DAPI onto clean microscopic glass slides.
- Place coverslips with cells facing down to allow contact between cells and DAPI solution.
NOTE: Cover slides with aluminum foil to protect from light. - Incubate for 30 min at room temperature.
- Store at -20 °C.
6. Confocal microscopy, image acquisition, and quantification using FIJI
NOTE: To acquire/capture immunofluorescence images, use a confocal laser scanning microscope. Oil-immersion 63x objective lens is recommended for optimal resolution.
- Allow coverslips to reach room temperature, protected from light, for at least 30 min prior to image acquisition.
- Clean the coverslips with absorbent tissue.
- Add a drop of immersion oil to the objective and place each slide under the microscope.
- Move the objective until the oil touches the slide.
- Observe and adjust the microscope focus; select the 63x objective with oil.
- Turn on lasers at 488 nm, 552 nm, and 405 nm wavelengths.
- Select image resolution: 1024 x 1024.
- Click on the Live button, set the Z stack, and press Begin. Repeat the process and then press End.
- After selecting the Maximum Projection option in the Tool menu, wait for the image to be acquired.
- Save the experiment.
- Export images in .lif format on a computer. Use FIJI open-source image analysis software to analyze images.
- Select the images to be analyzed and select Duplicate Image.
- To have the image in grayscale, select Image | Adjust | Color Threshold. Select 0 and 255 (saturation).
- Select the threshold method: Default.
- Select threshold color: B and W (black and white).
- Do not select Dark background.
- Select: Process | Binary | Options and then select the relevant data to be measured: Area, Min, Max, gray value, integrate density.
- Select the free hands' tool in the Fiji toolbar and trace each cell manually carefully.
- From the Analyze menu, select Set measurements. Make sure to have area integrated intensity and mean grey value selected. Repeat this process for each cell.
- Select all data in the Results window and copy and paste into a spreadsheet file.
- Calculate the corrected total cell fluorescence (CTCF): CTCF (Integrate density = Area of selected cell x Mean fluorescence of background readings).
- Open the file containing data using statistical analysis software to perform statistical analysis.
7. Statistical analysis
- Open a New Project when a welcome dialog appears.
- Choose the type of graph and medium with SD.
- Apply the values obtained from the experimental results to the table.
- Select Descriptive Statistics and choose the option column Statistics [all tests] to analyze data distribution.
- If data follow a Gaussian distribution, choose the t-test to analyze samples by comparing two pairs. If the distribution is non-Gaussian, analyze data using the Mann-Whitney test.
- Choose the best graph option for optimal data representation.
Representative Results
This protocol described herein enables the evaluation of cell migration and its associated mechanisms, such as actin dynamics and adhesion, thereby providing a tool to determine the migration of Leishmania-infected host cells within the vertebrate host. The results presented here demonstrate that this in vitro assay provides rapid and consistent indications of changes in cellular adhesion, migration, and actin dynamics prior to in vivo experimentation.
First, cells were successfully cultured following aseptic techniques and lab protocols. Data generated via migration analysis using cell culture membrane inserts allowed us to evaluate the migration of L. infantum-infected or uninfected human dendritic cells. DAPI staining permitted the facile visualization of migratory cells, enabling us to discriminate between infected and non-infected cells as staining procedures incorporate both dendritic cell and parasite nuclei. Infected cells can be identified by visualizing the large macrophage nuclei and the number of smaller amastigote nuclei clustered around each macrophage nucleus. Next, infected cells (L. infantum infected group) and uninfected cells (control group) were then counted for each field of vision using a manual counter. Finally, the number of migratory infected cells was compared to the number of uninfected cells (control group) that migrated. Our results indicate higher rates of cell migration following L. infantum infection when compared to non-infected controls (Figure 1).
The evaluation of actin dynamics and the formation of adhesion complexes, factors critical to cellular migration, allows for an enhanced understanding of how infection may modulate host cell migration. To assess these mechanisms, we performed immunostaining for molecules involved in actin dynamics (phalloidin, Rac1, Cdc42, and RhoA) and adhesion complex formation (FAK and paxillin). The expression of each protein was evaluated using confocal microscopy. The differences in protein expression were assessed by comparing the fluorescence intensity between infected and uninfected cells for each protein analyzed. Our results demonstrate actin polymerization in infected and non-infected cells and the formation and localization of adhesion complexes. DAPI staining enabled the identification of infected cells through the staining of parasite nuclei. Fluorescence analysis indicated increased FAK and paxillin expression in DCs following L. infantum infection (Figure 2). To evaluate the organization of actin filaments in DCs, actin was labeled with fluorescent phalloidin. The resulting images revealed more areas with actin polymerization, yet no differences in phalloidin staining comparing infected and non-infected cells (Figure 3A). However, considering that the structure of actin is highly dynamic, the evaluation of actin-associated molecules may provide additional insight into the organization of this structural protein. Thus, we evaluated the expression of Rho GTPase proteins. Although phalloidin staining yielded similar results in infected and non-infected cells, increases were noted in Rac1, Cdc42 and RhoA expression after L. infantum infection when compared to uninfected controls (Figure 3B,C,D). These results reinforce the need for further investigation of molecules involved in actin polymerization to gain a more comprehensive understanding of its associated dynamics.
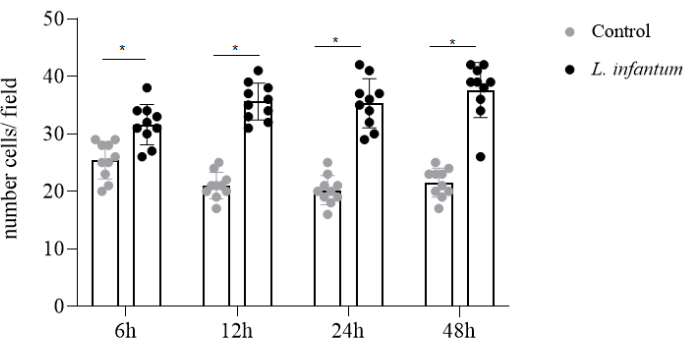
Figure 1: Evaluation of dendritic cell migration in L. infantum infection. Dendritic cells were infected by L. infantum at a ratio of 20:1 for 4 h. At 6, 12, 24 or 48 h after infection, dendritic cells could migrate in the presence of CCL3 chemoattractant through the cell culture insert system for an additional 4 h. Migrating cells were washed, fixed, and stained with DAPI. Bars represent numbers of migratory cells after L. infantum infection from random counts in 10 fields using confocal microscopy. Each dot represents one cell. *p<0.05 (Student's t-test). Please click here to view a larger version of this figure.
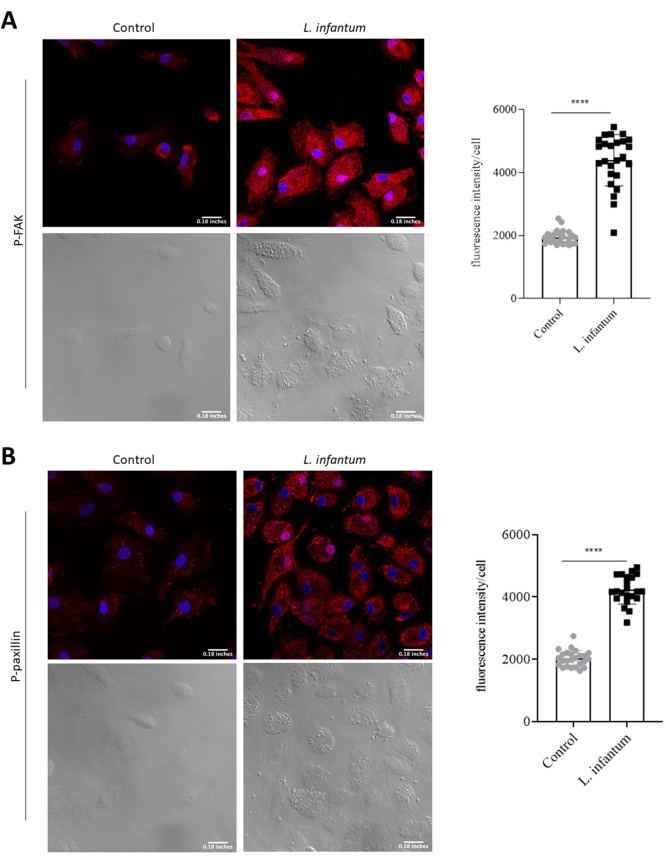
Figure 2: Evaluation of adhesion complex formation in L. infantum-infected dendritic cells. Dendritic cells infected or not with L. infantum were stained with anti-pFAK or anti-paxillin antibodies. (A) Fluorescence intensity of FAK expression. (B) Fluorescence intensity of paxillin expression. For each group, 30 cells were analyzed using FIJI software. Red: anti-pFAK, or anti-paxillin; Blue: DAPI; Grayscale: differential interference contrast (DIC). Scale bar = 0.18 inches. *p<0.05 (Student's t-test). Please click here to view a larger version of this figure.
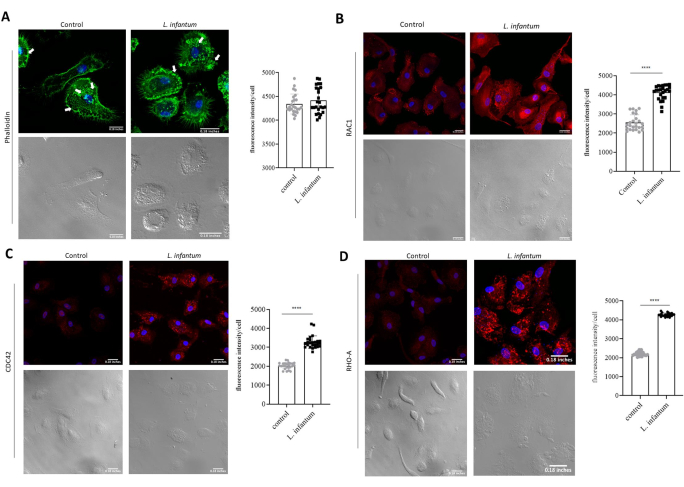
Figure 3: Evaluation of actin dynamics in L. infantum-infected dendritic cells. Dendritic cells infected or not with L. infantum were stained with anti-Rac1, anti-RhoA and anti-Cdc42 antibodies or fluorescent phalloidin. (A) Fluorescence intensity of phalloidin expression (green). (B) Fluorescence intensity of Rac1 expression (red). (C) Fluorescence intensity of Cdc42 expression (red) (D) Fluorescence intensity of RhoA expression (red). For each group, 30 cells were analyzed using FIJI software. Red: anti-Rac1 or anti-Cdc42; Green: anti-RhoA or phalloidin; Blue: DAPI; Grayscale: differential interference contrast (DIC). Scale bar = 0,18 inches. *, p<0,05 (Student's t-test). Please click here to view a larger version of this figure.
Discussion
The method described here for evaluating cell migration using the cell culture membrane inserts system allows researchers to study the migratory response of cells in a two-dimensional environment. In this technique, some steps are considered critical. Firstly, the differentiation of human DCs and infection with Leishmania are determinative since the infection rate is donor-dependent. Using more than one donor per experiment and healthy Leishmania cultures will allow for more consistent results. It is also crucial that parasites be maintained in host animals, which favors the selection and maintenance of virulent strains and the ability to readily colonize host cells. Following DC differentiation, we recommend checking the expression of surface markers CD80 and CD11c to verify that the cells being used in experimentation are, in fact, dendritic cells. Despite variability in infection rates, increased DC migration following L. infantum infection was observed in all experiments.
The cell culture membrane inserts system assay, employed herein, entails cell migration through a porous polycarbonate membrane from the upper to the lower compartment of the cell culture membrane inserts system. A chemoattractant is placed in the bottom of each well to direct cell migration through the porous membrane during incubation36. It is important to use an appropriate pore size for the cell type of interest. For DCs, we used a cell culture membrane inserts system with a 5μm pore size. Of note, after the fixation step, the surfaces of the insert membranes were scraped with a swab to remove cells that had not migrated. This step was implemented to ensure that only those cells that successfully migrated through the membrane were evaluated. Another critical point that warrants consideration is the use of DAPI staining, a rapid procedure that allows investigators to not only identify host cells but also parasite nuclei, thus enabling the convenient identification of infected cells.
Although the cell culture membrane inserts system has been extensively used as an effective tool for assessing migration28,36,37,38,39, there are some limitations associated with this technique. During cell washing and fixation steps, less-adherent cells may be lost when analyzing the membrane, resulting in an underestimation of the number of cells that migrated. Another limitation is time related. After long incubation periods, gradient loss may occur due to diffusion through the porous membrane. Thus, this system should be considered more efficient for shorter incubation periods36. On the other hand, the use of the cell culture membrane inserts system is advantageous compared to other methods such as the scratch assay or random migration since it allows the study of directional migration in the presence of a chemoattractant.
Another essential component of this protocol is the use of immunofluorescence to investigate mechanisms involved in cellular migration, such as adhesion complex formation and actin dynamics. This technique allows investigators to visualize specific targets in tissues or cells using specific antibodies for proteins of interest. In this protocol, to assess cellular adhesion, we evaluated the expression of phosphorylated FAK and paxillin, both crucial proteins involved in the formation of adhesion complexes in different cell types, including leukocytes13, and consequently excellent tools for studying cell adhesion. Previous studies have shown that increased FAK signaling promotes cellular motility11. Also, the use of this technique to evaluate cell adhesion does not require the use of more labor-intensive techniques, such as the use of inflamed connective tissue26.
The phalloidin staining technique40,41 provides information about the expression of polymerized actin, F-actin, and the regions of cells with higher polymerization rates. To gain further insight into actin dynamics in L. infantum-infected cells, we also evaluated the expression of Rac1, Cdc42, RhoA, and Rho GTPase, which participate in the polymerization of actin filaments42. The expression of these molecules was also performed using immunofluorescence. This technique is not only an alternative to the use of transgenic mice expressing fluorescent actin28 but also provides further information about molecules modulated during the actin polymerization process.
Several factors can affect immunofluorescence quality and efficacy. Antibody dilution, for example, when not carefully determined, can impair the acquisition of images and lead to non-specific staining due to elevated levels of background signals. Samples must also always be protected from light to avoid any loss of cell fluorescence or staining43.
In summary, here we describe an in vitro protocol that allows for the evaluation of cell adhesion and migration processes in the context of Leishmania infection. Our primary focus was on DCs, which are known to play a significant role in the immunopathogenesis of leishmaniasis; however, how these cells participate in parasite dissemination in the vertebrate host remains poorly understood. This protocol can also be modified to investigate cellular migration in other types of host cells for the investigation of other species of intracellular parasites. Also, extracellular matrix, such as collagen I or Matrigel, can be added to the cell culture membrane inserts system to evaluate 3D migration and cellular invasion in different fields of study.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by Bahia Research Support Foundation (Fapesb), grant number 9092/2015. The authors acknowledge CNPq, Capes and Fapesb for financial support via scholarships. The authors would like to thank Andris K. Walter for critical analysis, English language revision and manuscript copyediting assistance.
Materials
| 16 Gauge needle | Descarpack | 353101 | |
| 24 well cell culture plate | JET-BIOFIL | J011024 | |
| 25 Gauge needle | Descarpack | 353601 | |
| Albumin from bovine serum | Sigma Aldrich | A2153-100G | |
| Ammonium chloride | Sigma Aldrich | A-0171 | |
| Anti-mouse IgG, Alexa Fluor 488 | Invitrogen | A32723 | |
| Anti-mouse IgG, Alexa Fluor 594 | Invitrogen | A11032 | |
| Anti-rabbit IgG, Alexa Fluor 568 | Invitrogen | A11011 | |
| CD14 MicroBeads | MACS Myltenyi Biotec | 130-050-201 | |
| Cell Culture Flask 25cm2 | SPL | 70125 | |
| Cellstripper | Corning | 25-056-CI | |
| Confocal microscope | Leica | TCS SP8 | |
| Coverslip circles 13mm | Perfecta | 10210013CE | |
| Dissecting Forceps | VWR | 82027-406 | |
| EDTA | Sigma Aldrich | E6758 | |
| Falcon tube | KASVI | K19-0051 | |
| Fetal Bovine Serum | gibco | 16000044 | |
| Fluorescence microscope | Olympus | BX51 | |
| Glass slide 25,4×76,2mm | Perfecta | 200 | |
| Hemin bovine | Sigma Aldrich | H2250 | |
| Hemocytometer | Perfecta | 7302HD | |
| Histopaque® 1077 | Sigma Aldrich | 10771 | |
| MACS buffer | MACS Myltenyi Biotec | 130-091-221 | |
| Minimum Essential Medium | Gibco | 41090093 | |
| Mouse anti-Rac1 | BD | 610650 | |
| Paraformaldehyde | Sigma Aldrich | 158127 | |
| Phalloidin Alexa Fluor 488 | Invitrogen | A12379 | |
| Phosphate Buffered Saline | ThermoFisher | AM9624 | |
| Polycarbonate Membrane Transwell Inserts – Pore size 5.0 µm | Corning | 3421 | |
| ProLong Gold DAPI kit | Invitrogen | P36931 | |
| Rabbit anti-Cdc42 | Invitrogen | PA1-092X | |
| Rabbit anti-FAK (pTyr397) | Invitrogen | RC222574 | |
| Rabbit anti-paxilin (pTyr118) | Invitrogen | QF221230 | |
| Rabbit anti-RhoA | Invitrogen | OSR00266W | |
| Recombinant Human CCL3 | R&D Systems | 270-LD-010 | |
| Recombinant Human GM-CSF | PeproTech | 300-03 | |
| Recombinant Human IL-4 | PeproTech | 200-04 | |
| Recombinant Human M-CSF | PeproTech | 300-25 | |
| RPMI 1640 Medium | Gibco | 21870076 | |
| Saponin | Sigma Aldrich | 47036 – 50G – F | |
| Syringe 3 mL | Descarpack | 324201 | |
| Trypan Blue | Gibco | 15250061 |
References
- Burza, S., Croft, S. L., Boelaert, M. Leishmaniasis. The Lancet. 392 (10151), 951-970 (2018).
- Bi, K., Chen, Y., Zhao, S., Kuang, Y., John Wu, C. H. Current Visceral Leishmaniasis Research: A Research Review to Inspire Future Study. BioMed Research International. 2018, (2018).
- Serafim, T. D., Iniguez, E., Oliveira, F. Leishmania infantum. Trends in Parasitology. 36 (1), 80-81 (2020).
- Van Assche, T., Deschacht, M., da Luz, R. A. I., Maes, L., Cos, P. Leishmania-macrophage interactions: Insights into the redox biology. Free Radical Biology and Medicine. 51 (2), 337-351 (2011).
- Podinovskaia, M., Descoteaux, A. Leishmania and the macrophage: A multifaceted interaction. Future Microbiology. 10 (1), 111-129 (2015).
- Antoine, J. C., Prina, E., Jouanne, C., Bongrand, P. Parasitophorous vacuoles of Leishmania amazonensis-infected macrophages maintain an acidic pH. Infection and Immunity. 58 (3), (1990).
- Antoine, J. C., Prina, E., Lang, T., Courret, N. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends in Microbiology. 6 (10), 392-401 (1998).
- Friedl, P., Wolf, K. Plasticity of cell migration: A multiscale tuning model. Journal of Cell Biology. 188 (1), 11-19 (2010).
- Sheetz, M. P., Felsenfeld, D., Galbraith, C. G., Choquet, D. Cell migration as a five-step cycle. Biochemical Society symposium. 65, 233-243 (1999).
- Yano, H., et al. Roles played by a subset of integrin signaling molecules in cadherin-based cell-cell adhesion. Journal of Cell Biology. 166 (2), 283-295 (2004).
- Mitra, S. K., Hanson, D. A., Schlaepfer, D. D. Focal adhesion kinase: In command and control of cell motility. Nature Reviews Molecular Cell Biology. 6 (1), 56-68 (2005).
- Deramaudt, T. B., et al. Altering FAK-Paxillin Interactions Reduces Adhesion, Migration and Invasion Processes. PLoS One. 9 (3), (2014).
- Turner, C. E. Paxillin and focal adhesion signalling. Nature Cell Biology. 2 (12), (2000).
- Linder, S., Aepfelbacher, M. Podosomes: Adhesion hot spots of invasive cells. Trends in Cell Biology. 13 (7), 376-385 (2003).
- Huveneers, S., Danen, E. H. J. Adhesion signaling – Crosstalk between integrins, Src and Rho. Journal of Cell Science. 122 (8), 1059-1069 (2009).
- Jones, G. E. Cellular signaling in macrophage migration and chemotaxis. Journal of Leukocyte Biology. 68 (5), 593-602 (2000).
- De Fougerolles, A. R., Koteliansky, V. E. Regulation of monocyte gene expression by the extracellular matrix and its functional implications. Immunological Reviews. 186 (1), 208-220 (2002).
- Cortesio, C. L., Boateng, L. R., Piazza, T. M., Bennin, D. a., Huttenlocher, A. Calpain-mediated proteolysis of paxillin negatively regulates focal adhesion dynamics and cell migration. The Journal of Biological Chemistry. 286 (12), 9998-10006 (2011).
- Verkhovsky, A. B., et al. Orientational order of the lamellipodial actin network as demonstrated in living motile cells. Molecular Biology of the Cell. 14 (11), 4667-4675 (2003).
- Ridley, A. J., et al. Cell Migration: Integrating signals from front to back. Science. 302 (5651), 1704-1709 (2003).
- Hall, A. Small GTP-binding proteins, and the regulation of the actin cytoskeleton. Annual Review of Cell Biology. 10, 31-54 (1994).
- Machesky, L. M., Hall, A. Rho: A connection between membrane receptor signalling and the cytoskeleton. Trends in Cell Biology. 6 (8), 304-310 (1996).
- Ridley, A. J., Hall, A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 70 (3), 389-399 (1992).
- DeMali, K. A., Burridge, K. Coupling membrane protrusion and cell adhesion. Journal of Cell Science. 116 (12), 2389-2397 (2003).
- López-Colomé, A. M., Lee-Rivera, I., Benavides-Hidalgo, R., López, E. Paxillin: A crossroad in pathological cell migration. Journal of Hematology and Oncology. 10 (1), (2017).
- Carvalhal, D. G. F., et al. The modelling of mononuclear phagocyte-connective tissue adhesion in vitro: Application to disclose a specific inhibitory effect of Leishmania infection. Experimental Parasitology. 107 (3-4), 189-199 (2004).
- Pinheiro, N. F., et al. Leishmania infection impairs β1-integrin function and chemokine receptor expression in mononuclear phagocytes. Infection and Immunity. 74 (7), 3912-3921 (2006).
- de Menezes, J. P. B., et al. Leishmania infection inhibits macrophage motility by altering F-actin dynamics and the expression of adhesion complex proteins. Cellular Microbiology. 19 (3), (2017).
- Hermida, M. D. R., Doria, P. G., Taguchi, A. M. P., Mengel, J. O., dos-Santos, W. L. C. Leishmania amazonensis infection impairs dendritic cell migration from the inflammatory site to the draining lymph node. BMC Infectious Diseases. 14 (1), 450 (2014).
- Ballet, R., et al. Blocking junctional adhesion molecule c enhances dendritic cell migration and boosts the immune responses against Leishmania major. PLoS Pathogens. 10 (12), (2014).
- Rocha, M. I., et al. Leishmania infantum enhances migration of macrophages via a phosphoinositide 3-Kinase î-dependent pathway. ACS Infectious Diseases. 6 (7), 1643-1649 (2020).
- Hiasa, M., et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-α converting enzyme (TACE). Blood. 114 (20), 4517-4526 (2009).
- de Melo, C. V. B., et al. Phenotypical characterization of spleen remodeling in murine experimental visceral leishmaniasis. Frontiers in Immunology. 11, 653 (2020).
- Gibaldi, D., et al. CCL3/macrophage inflammatory protein-1α is dually involved in parasite persistence and induction of a TNF- and IFNγ-enriched inflammatory milieu in Trypanosoma cruzi-induced chronic cardiomyopathy. Frontiers in Immunology. 11, 306 (2020).
- Finger, P. T., Papp, C., Latkany, P., Kurli, M., Iacob, C. E. Anterior chamber paracentesis cytology (cytospin technique) for the diagnosis of intraocular lymphoma. British Journal of Ophthalmology. 90 (6), 690-692 (2006).
- Zhang, C., Barrios, M. P., Alani, R. M., Cabodi, M., Wong, J. Y. A microfluidic Transwell to study chemotaxis. Experimental Cell Research. 342 (2), 159-165 (2016).
- Glynn, S. A., O’Sullivan, D., Eustace, A. J., Clynes, M., O’Donovan, N. The 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors, simvastatin, lovastatin and mevastatin inhibit proliferation and invasion of melanoma cells. BMC Cancer. 8, (2008).
- van de Merbel, A. F., vander Horst, G., Buijs, J. T., vander Pluijm, G. Protocols for migration and invasion studies in prostate cancer. Methods in Molecular Biology. 1786, 67-79 (2018).
- Omar Zaki, S. S., Kanesan, L., Leong, M. Y. D., Vidyadaran, S. The influence of serum-supplemented culture media in a transwell migration assay. Cell Biology International. 43 (10), 1201-1204 (2019).
- Borovikov, I. S. Izuchenie strukturnykh izmeneniǐ sokratitel’nykh belkov myshechnogo volokna s pomoshch’iu poliarizatsionnoǐ ul’trafioletovoǐ fluorestsentnoǐ mikroskopii. VIII. Vliianie glutaral’degida i falloidina na konformatsiiu F-aktina. Tsitologiya. 26 (11), 1262-1266 (1984).
- Cooper, J. A. Effects of cytochalasin and phalloidin on actin. The Journal of Cell Biology. 105 (4), 1473-1478 (1987).
- Allen, W. E., Jones, G. E., Pollard, J. W., Ridley, A. J. Rho, Rac and Cdc42 regulate actin organization and cell adhesion in macrophages. Journal of Cell Science. 110, 707-720 (1997).
- Lichtman, J. W., Conchello, J. A. Fluorescence microscopy. Nature Methods. 2 (12), 910-919 (2005).

.