In order to capture the regional Ca2+ and brightfield information for the entire length of the trabecula presented here, seven muscle positions were required. Figure 6 suggests that the twitch force was undisturbed by this motion, revealing that there was no position dependence of the active force production.
B-scans collected using optical coherence tomography at a rate of 100 Hz were segmented using the ImageJ plugin WEKA14 (Figure 7A). Each cross-section appears distorted due to the difference between the lateral (10 µm) and depth (1.73 µm (in myocardium)) resolutions. This distortion was corrected by scaling the image's depth axis by the lateral resolution-depth resolution ratio. Figure 7B,C demonstrate that after scaling the raw C-scan of the trabecula it is approximately cylindrical in geometry. The reflection of the measurement chamber wall can sometimes overlap with the muscle data (Figure 7A,B), but the segmentation software can be trained to account for this (Figure 7D,E). Once segmented, the cross-sectional area along the length of the muscle can be calculated throughout the twitch (Figure 7F). Note that this particular trabecula has a small appendage branching from it. The motion of the branch is evident ~0.75 mm along the trabecula. Finally, the segmented images can be converted into meshes to aid the construction of geometric models (Figure 7G).
Imaging data captured at each of the different trabecula positions at a rate of 100 fps were stitched together to create a single complete image of the trabecula (Figure 8A). The resolution of these images is 0.535 µm/pixel. The use of linear weighting functions in the overlapping regions of neighboring windows aids visualization and minimizes the impact of the vignette present in the brightfield images. To measure the fluorescent signal, a 540 µm by 540 µm window of the trabecula is cyclically illuminated with 340 nm, 365 nm, and 380 nm wavelength light at a rate of 600 Hz. The ratio of the emitted fluorescence associated with the 340 nm and 380 nm excitation light correlates to the intracellular calcium after the trabecula has been loaded with Fura-2. As this measurement is a ratio, the effective measurement rate is 200 Hz. The averaged (n = 10) intracellular Ca2+ transients from each window are aligned with the region they were imaged (Figure 8B). While the peak of the transients appear reasonably consistent, the diastolic [Ca2+] is lower within the region between 900 µm and 1800 µm along the trabecula. Similarly, the results of the displacement tracking (Figure 8C) and sarcomere length (Figure 8D) calculations also indicate the presence of regional variability. The markerless tracking technique used is able to process the displacement of each pixel, given enough contrast. When mapping the distribution of sarcomere lengths in post, a cross-correlation area of 128 pixels by 128 pixels (~67 µm by 67 µm) was used to calculate regional sarcomere length. This area encapsulates approximately 29 sarcomeres when close the sample is close to optimal sarcomere length. The step-size (in both the x- and y-directions) between the centroid of each cross-correlation window was set to 50 pixels (~26 µm) for processing these data. The suitability of sarcomere length estimates were tested based on the width and amplitude of the Gaussian fit to the FFT signal. These conditions were not met in the muscle region between 0 µm and 500 µm so no sarcomere length information could be computed there. Given the associated displacements, it is likely that the sarcomeres in this region elongated during the contractile phase. In keeping with this speculation, the average sarcomere lengths on the right-hand side of the trabecula shorten during that period. By combining the information provided by each of the panels, it appears that the region with the largest cross-sectional area does not produce the most significant force. On the assumption that the regional variation of Ca2+ transients has an approximately smooth gradient, Figure 8B indicates that the largest amplitude Ca2+ transient occurs somewhere between 1300 µm and 1600 µm along the trabecula. The displacement map indicates that the region that undergoes the least motion aligns well with the peak Ca2+ transient. However, this region has the smallest cross-sectional areas of the sample. With these data in mind, one could infer that this region generated the most stress.

Figure 1: Annotated image of the cardiomyometer. Each of the main optical components is outlined. The inset contains a close-up, rear-view, of the microscope objective in situ, below the measurement chamber. Please click here to view a larger version of this figure.
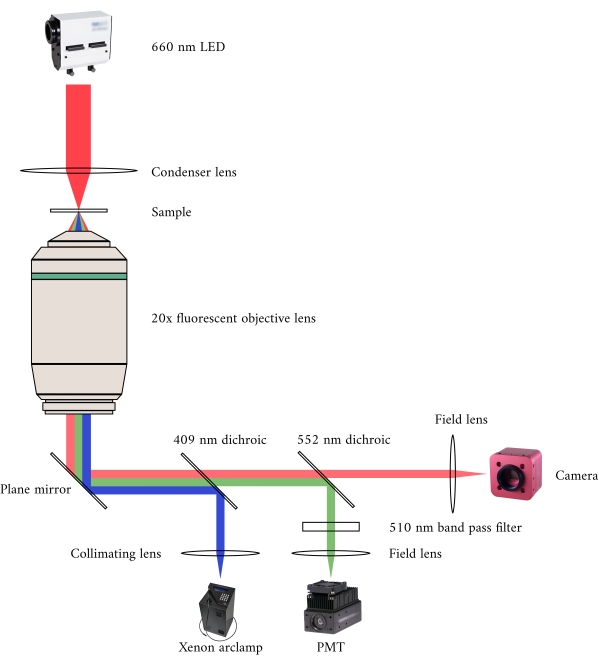
Figure 2: Optical pathway for simultaneous brightfield and fluorescence microscopy. The illumination source for the fluorescence microscope is a Xenon arc lamp, the output of which cyclically switches between 340 nm, 365 nm, and 380 nm light. The arc lamp output path contains a dichroic mirror with a cut-off wavelength of 409 nm that reflects the UV light onto a mirror that directs the light into a fluorescence microscope objective. The lens focuses the excitation light onto the sample and collects the emitted light, which has a longer wavelength of 510 nm. This emitted light passes through the first dichroic mirror, but not the second, as it has a cut-off wavelength of 552 nm. A field lens then focuses the reflected light onto the sensor of the PMT. Meanwhile, the illumination source (660 nm LED) for the brightfield microscope is located above the sample. The transmitted light is focused onto the sample by a condenser lens, and the 20× fluorescence objective captures the resultant transmission image. The wavelength used for brightfield illumination exceeds the cut-off wavelength of each dichroic mirror, so it passes through both of them before the image is focused onto the sensor of a CMOS camera. Please click here to view a larger version of this figure.
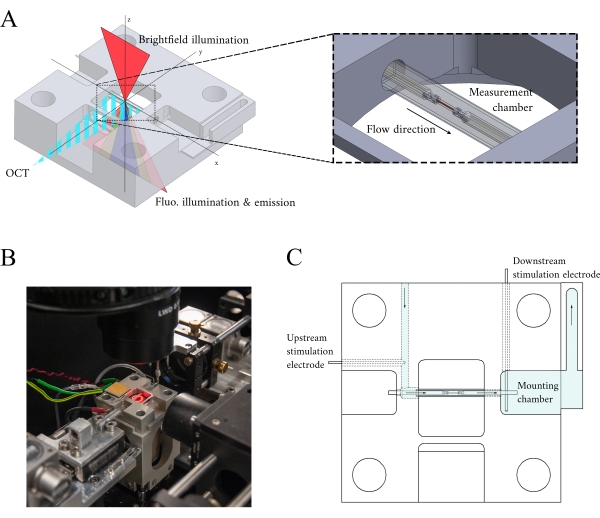
Figure 3: Measurement chamber holder design. (A) Isometric view of the measurement chamber holder with optical paths overlaid. Brightfield illumination occurs from the superior surface (z-axis); fluorescence illumination occurs from the inferior surface (z-axis), and the measurement arm OCT signal is orthogonal to the other illumination axis (y-axis). During the experiment, two platinum hooks hold a trabecula within a glass capillary tube that functions as the measurement chamber. Voice-coil motors control each hook and their positions are measured using laser interferometry. The current position is compared with a user-defined set point and, using a PID controller encoded within an FPGA, the error is minimized. (B) Measurement chamber in situ with the brightfield illumination turned on. The back-view is shown in the Figure 1 inset. (C) Schematic of the superfusate flow through the measurement chamber holder. Superfusate enters the rear of the block and flows in the direction indicated by arrows. The upstream and downstream electrodes establish the field stimulation for eliciting the contraction of a mounted trabecula in the measurement chamber. Blue shading indicates the regions where the superfusate flows during an experiment. Please click here to view a larger version of this figure.
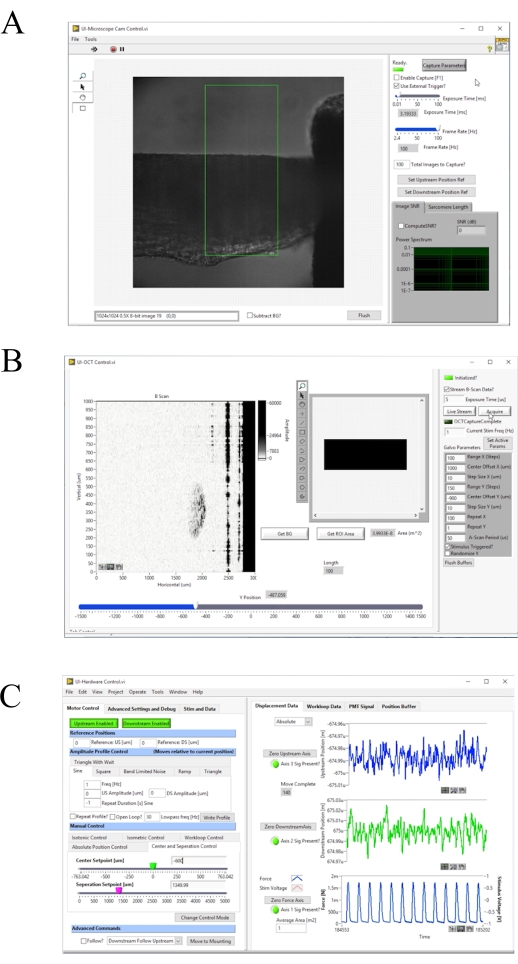
Figure 4: Front panel of the image acquisition and control software. (A) Brightfield imaging user interface. (B) OCT imaging user interface. (C) Hardware control user interface. Please click here to view a larger version of this figure.
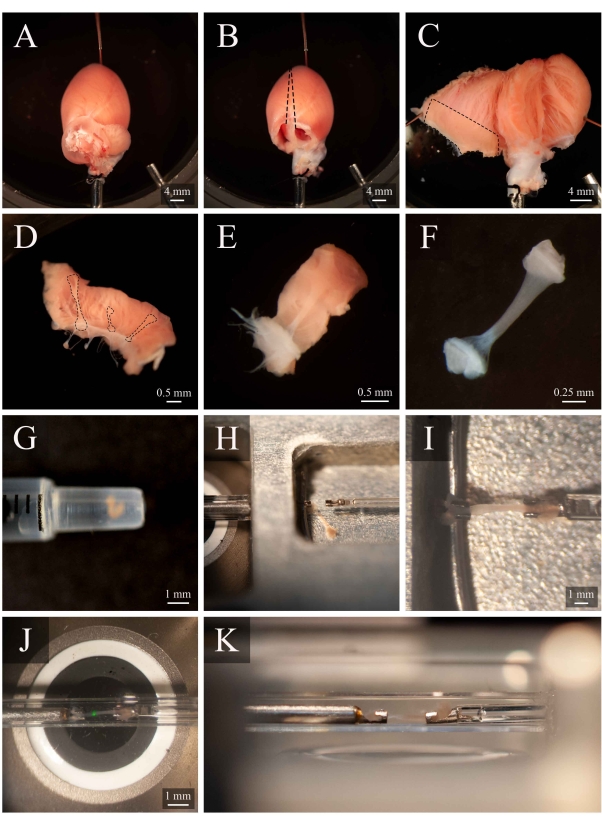
Figure 5: Trabecula dissection and mounting protocol. (A) Langendorff-perfused rat heart in the dissection chamber. (B) The same heart with the atria removed. Dashed lines indicate the excision trajectory to open the ventricles. (C) An opened heart to expose the interior of both ventricles. The dashed box indicates the region where trabeculae are typically found. (D) Excised right-ventricular wall region (the same as indicated by the dashed box in C). Dashed lines highlight three trabeculae. (E) A trabecula selected from the three in D. (F) The trabecula from panel E with the wall tissue removed. (G) The isolated trabecula in the end of a 1 mL syringe. (H) The trabecula in the mounting chamber. (I) The trabecula mounted between two platinum hooks. (J) The trabecula, mounted between hooks, within the measurement chamber (Figure 3B). The green spot is an artefact from the first dichroic filter. (K) Secondary angle of the mounted trabecula within the measurement chamber. The distance between the trabecula and the microscope objective lens is approximately 1 mm. Please click here to view a larger version of this figure.
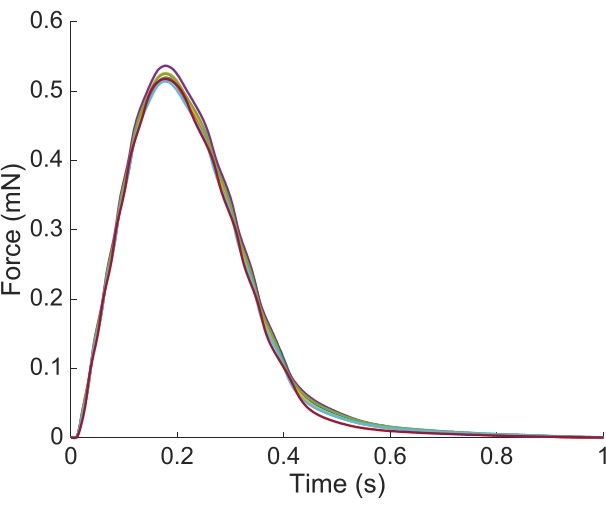
Figure 6: Position dependence of the force measurement. The force produced by the muscle from each of the imaging positions (n = 7) overlaid. The average active force production was 0.527 mN ± 0.003 mN, time to 50 % contraction 77.1 ms ± 0.3 ms, and time to 50 % relaxation 328.1 ms ± 0.9 ms (all data are presented as the mean ± SE). Please click here to view a larger version of this figure.
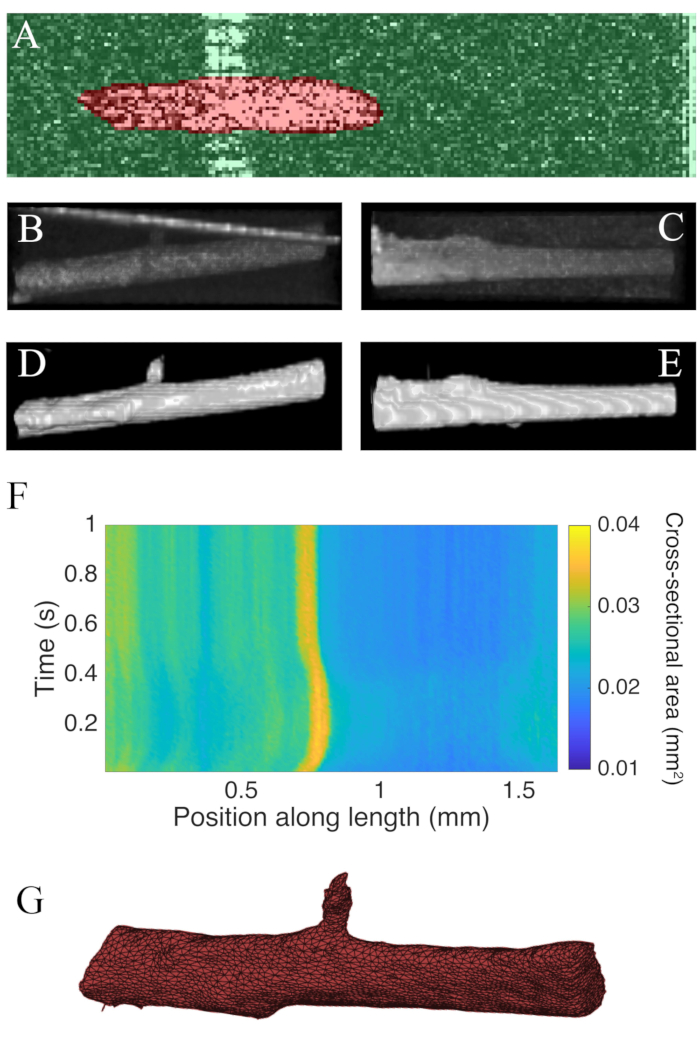
Figure 7: OCT imaging analysis. (A) Example of WEKA segmentation. The segmented cross-section of the muscle is highlighted in red, and the background is highlighted in green. (B) Superior view of the raw C-scan data of a trabecula. The bright angled line towards the top of the image is the reflection of the measurement chamber wall. (C) Lateral view of the raw C-scan data of a trabecula. (D) Superior view of the segmented OCT data. (E) Lateral view of the segmented OCT data. (F) The cross-sectional area along the length of the trabecula (x-axis) through time (y-axis). The average cross-sectional area along the muscle length was 0.0326 mm2 ± 0.0005 mm2 (mean ± S.E.) (G) A mesh of the trabecula. The mesh has been approximately aligned with the cross-sectional area plot of panel F. Please click here to view a larger version of this figure.
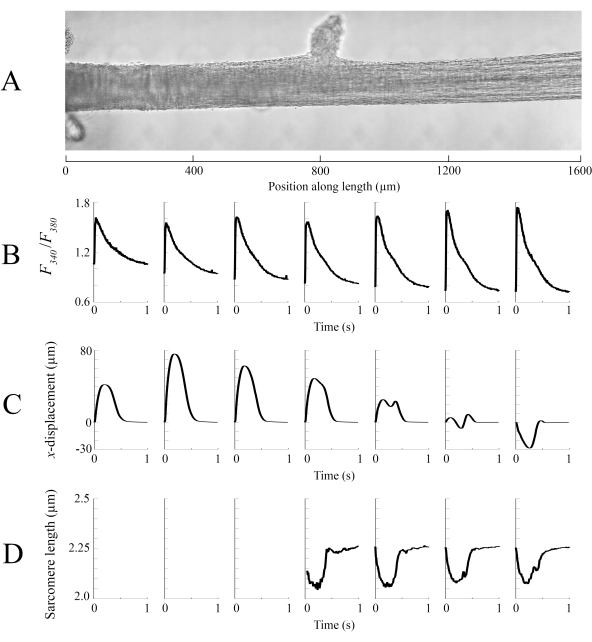
Figure 8: Brightfield and fluorescence imaging analysis. (A) Stitched image (seven imaging windows) of the trabecula. (B) Ca2+ transients along the length of the trabecula. (C) Average x-displacement from each imaging window. A positive displacement represents a motion to the right and negative to the left. (D) Average sarcomere lengths from each imaging window that had the necessary image contrast. Please click here to view a larger version of this figure.
Table 1: Table of Solutions Please click here to download this Table.
