Microinjection of trapping beads:
Microspheres injected into the one-cell zebrafish embryo spread over the entire animal cap during morphogenesis. For a clearer visualization, we repeated the injection protocol with red fluorescent microbeads and took volumetric images with our confocal microscope at different developmental stages. In Figure 4A–D, injected beads are visualized in the cytoplasm of progenitor stem cells in vivo at 5 hfp. Later on, microspheres appeared spread over the whole embryo at 24 hpf (Figure 4E). Embryos at both stages developed normally and survival rates were comparable with control non-injected or mock-injected embryos (see Figure S3). This is consistent with other studies that report unperturbed survival of bead-injected zebrafish up to 5 days post fertilization55.
Our spinning-disk confocal microscope is compatible with multi-channel fluorescence microcopy. In Figure 5A, we show isolated stem cells with one or two beads in the cytoplasm. Multiple fluorescent labels can be used to investigate different aspects of the cell (Figure 5B). Nuclear morphology can be tracked with a Hoechst dye or using a H2A::mCherry mRNA expression, while inner nuclear membrane can be analyzed with Lap2b-eGFP12. Dynamics of the actomyosin cortex, as well as intracellular calcium levels, can be observed with a My12.1::eGFP transgenic line56 and Calbryte-520 incubation, respectively. The protocol that has been described here aims to compare cell nucleus mechanics of immobilized wildtype cells on adhesive substrates (later referred to as suspension) and in mechanical confinement. Isolated stem cells confined in microchambers of 10 µm height exhibited partial unfolding of the inner nuclear membrane (INM) and a subsequent increase in actomyosin contractility12. In Figure 5C, confined cells with one or two beads in the cytoplasm are shown. Successful confinement will be visible via flattened, expanded cells with a wider cross section of the nucleus. The nuclear membrane is further unfolded in confined cells and should appear smoothened out in comparison to cells in suspension (Figure 5C).
Force-time and force-deformation analysis
The analysis of the obtained results strongly depends on the investigated specimen and the question of interest and thus they cannot be generalized here. As an example, a common way to analyze indentation measurement is to extract a Young's modulus by fitting a modified Hertz model to the force-indentation data57. However, the assumption for such a treatment needs to be carefully assessed and might not always be properly justified (such as the investigated structure being isotropic, homogenous, with linear elasticity and indentations being smaller than the bead radius). We thus only consider model independent measurements here that allow the mechanical behavior of the investigated structure to be compared among different experimental scenarios.
As a starting point, measuring the slope of the force-displacement curve at a certain indentation depth provides a measure of a model independent structural stiffness58 of the nucleus. This value can then be collected from multiple samples and compared between varying experimental settings and sample perturbations.
Indentation measurement
In the following lines, we focus on the mechanical response of the cell nucleus during cell deformation in confinement. Experiments in step 8 of this protocol typically lead to force peaks of up to 200 pN for indentation depths of approximately 2-3 µm. However, these values can be largely different, depending on the cell type and experimental conditions, with softer nuclei leading to lower force for a given indentation. It is thereby needed to accurately measure the nuclear deformation, together with force, for an accurate mechanical characterization of the cell nucleus. In this section, we will obtain the cell nuclear stiffness from representative force indentation measurements.
In Figure 6, we show the deformations of the distal and proximal sides of a nucleus in a suspended and confined cell. A rich mechanical behavior can be observed. In a typical suspended cell on an adhesive substrate, the nucleus was strongly indented by the bead, but also slightly displaced upon repetitive pushing events. We measured the bead indentation onto the nucleus by analyzing the kymographs obtained from fluorescence imaging of Hoechst-stained cell nuclei. Kymographs were easily computed using Fiji's Multi Kymograph plugin along the indentation direction (Figure 6A,B) and imported into Matlab (Version 2021, Mathworks) for further processing. A step function was fitted to the raw intensity profile with the aim to track the delimiting edges of the nucleus along the trajectory of the indentation routine. As can be seen, it bears accurate information on the nuclear change in shape (Figure 6 and Figure S2). We used the following double-sigmoid curve as an analytical version of a step function:
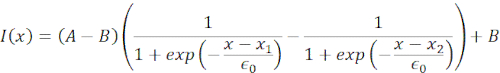 (Equation 1)
(Equation 1)
Here, x1 and x2 denote the distal and proximal edges of the nucleus, while A and B are the maximum and background gray values of the blue channel (Hoechst dye) of the image (Figure 6B). The edge width has been considered (e0 = 0.25 mm). While the indented, proximal nucleus edge (x2) followed the trajectory applied by the optical trap routine after the microsphere-nucleus contact, the opposite, distal edge (x1) displays relaxation dynamics as expected for a viscoelastic material such as the cytoplasm (Figure 6D). In contrast, nuclei in cells confined in 10 µm high microchambers do not exhibit such translocation behavior of the nucleus upon indentation within the cell (Figure 6B,D). Also shown in Figure 6D, the rear edges of the nuclei remains unaltered by the bead pushing from the proximal side, most likely due to stronger forces arising from cell contractility and friction acting against the indentation force. In order to get the correct deformation depth, the displacement x1 was subtracted from the indented measure x2: Δx = x2 – x1 (see also Figure 6D).
Force data analysis
The force causing nuclear deformation was measured from the change in light momentum originated at the optically-trapped microbead (Figure 7A). The force upon applying trapezoidal trajectories (step 8.4.3, Figure 7B) initially increased linearly until the trap stopped moving, but then relaxed to a steady state value. This behavior indicated a viscoelastic material exhibiting loss and storage moduli. Right after the indentation event, the force reached a peak value, Fp, followed by a stress relaxation (Figure 7C):
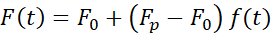 (Equation 2)
(Equation 2)
where F0 is the stored force for the elastic component and f(t) is a dimension-less relaxation function. We have analyzed this behavior in three ways:
1. Considering a standard linear solid with an exponential stress relaxation, i.e., f(t) = e–t/τ, schematically represented in Figure 7C inset.
2. Using a general, double-exponential decay:
F(t) = A + B1e-t/τ1 + B2e-t/τ2.
3. Using a power law followed by an exponential decay59:
f(t) = t-pe-t/τ, fitted in Figure 7C.
While the fit for model 1 can be carried out straightforwardly, we recommend to estimate the initial guesses for (τ1, τ2) and (p, τ) for models 2 and 3, respectively. This can be performed, respectively, by fitting lines onto the data in logarithmic-versus-linear (Figure 7D, left) and logarithmic-versus-logarithmic (Figure 7D, right) scales. Table S3 summarizes the results for the example analyzed in Figure 7. In the following section, we will consider the combination of a power law and an exponential law for the characterization of the cell nucleus mechanics.
Force displacement relation
Likewise, the described experimental set-up can be used to obtain the force-displacement relation of multiple indentation events. By performing triangular routines (step 8.4.4, Figure 8A), it is possible to relate the force to the deformation and plot a force-indentation curve. An exemplary outcome is shown in Figure 8B, in which a flat baseline smoothly changed slope once the bead got into contact with the nucleus. Identifying the true contact point in the noisy data is a challenge, and care has to be taken to see whether the contact region is fit to elastic models60. In this particular experiment, it could also be seen that the subsequent indentations result in curves with deeper contact points, indicative for too slow nuclear shape recovery after bead retraction and a change in the hysteretic cycle defined by the nucleus viscoelastic material properties (Figure 8C). Thus, the researcher should be aware if this happens and incorporate this into the analytical pipeline, or restrict the number of subsequent measurements such that this effect does not modify the measurement.
Nucleus mechanics in cells in suspension and under 10 µm confinement
The aforementioned approach was used to analyze the dynamics of nucleus stress relaxation in suspended cells on adhesive substrates and confined cells. Our results show that the confinement results in an expansion of the projected area (Figure 9A), but insignificant change in nuclear stiffness (Figure 9B). We measured similar relaxation with τ = 6.08 ± 1.1 s (unconfined) and τ = 4.00 ± 0.6 s (confinement), which indicates fast viscoelastic dissipation, followed by a stored force value that corresponds to the elastic modulus of the nucleus. In order to account for experimental variations, which may be produced by different initial conditions in the indentation routines, measured stored forces were normalized to the indentation depth, as 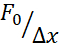 . This parameter accounts for the nucleus stiffness and describes the force, or the stress, necessary for a certain indentation. We obtained similar stiffness under confinement and in unconfined cells: = 20.1 ± 12.6 pN/µm and = 24.6 ± 13.6 pN/µm (mean ± standard deviation), respectively.
. This parameter accounts for the nucleus stiffness and describes the force, or the stress, necessary for a certain indentation. We obtained similar stiffness under confinement and in unconfined cells: = 20.1 ± 12.6 pN/µm and = 24.6 ± 13.6 pN/µm (mean ± standard deviation), respectively.
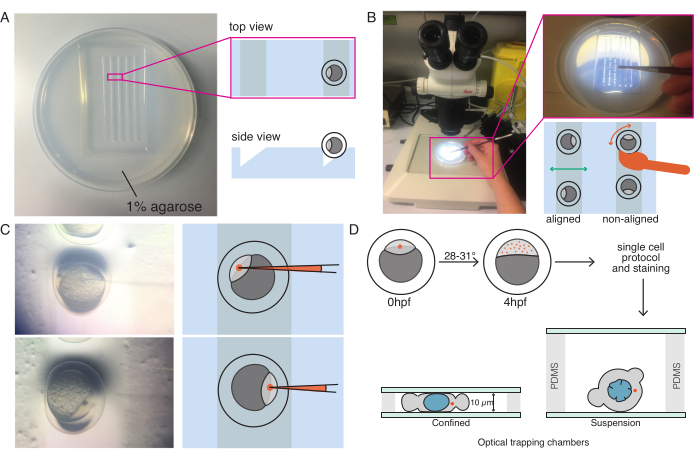
Figure 1: Microinjection of zebrafish embryos at one-cell (zygote) stage. (A) Injection plate: a triangular-shape injection plate is used for the injection. The plate is made of 1% ultrapure agarose in E3 (Egg's medium). Top and side views are shown on the right. (B) Embryo positioning: gently orient the embryos using a brush and orient such that the one-cell is clearly visible and easily accessible with the needle. We suggest to orient the embryos with the cell located in the opposite side of the needle, as shown in the sketch. (C) Injection procedure into the one-cell stage embryo: pierce the chorion surrounding the embryo and the single cell with the needle. Be sure that the tip of the needle is inside the cell and release the pressure to inject. (D) Incubate the embryos at 28-31 °C until they develop up to the blastula (sphere) stage (4 hpf). Perform the cell isolation protocol and cell staining (step 2) and prepare the optical trapping chamber with isolated cells in suspension and/or confinement combined with the corresponding substrate surface coating (step 3). Please click here to view a larger version of this figure.
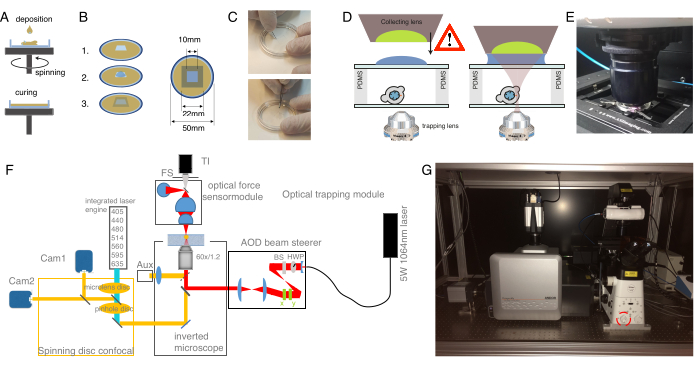
Figure 2: Preparation of the optical tweezer apparatus. (A) Spin-coating layers of PDMS with a defined height onto glass bottom dishes. The PDMS drop will spread out evenly due to the centrifugal force. (B) Preparation of the sample chamber out of the PDMS layer. 1: cut a square with a scalpel, 2: coat the inner well with concanavalin A (ConA), wash and seed cells; 3: cover with a glass slide or cover slip to seal the well. (C) Picture of the square cutting with a scalpel and removing the PDMS well with forceps. (D) Mounting the collecting lens of the optical force sensor over the trapping chamber. A drop of immersion oil serves as an immersion medium between the collecting lens and the upper glass cover. Schematic not to scale. Be cautious while lowering the collecting lens to not touch the glass cover of the sample dish. (E) Picture of the force detection unit in contact with the sample. (F) Schematic of the experimental set-up. The optical micromanipulation module uses a continuous wave laser beam (5W, λ = 1064 nm) with power control through a half-wave plate (HWP) and a polarizing beam splitter (BS). After being modulated with a pair of AODs, it is coupled to the upper epifluorescence port of an inverted microscope. The laser beam is then reflected by a 950 nm short-pass dichroic mirror (IR-DM), allowing for transmittance of fluorescence excitation and emission. The trapping laser is guided into the rear, epifluorescence port of the microscope (upper turret). The OTs are created at the focal plane of a water-immersion objective lens (60x, NA = 1.2). The optical force sensor is subjected by the microscope turret and captures the laser light emerging from the OTs with a high-NA, oil-immersion lens. At the same time, the force sensor enables bright-field illumination. The spinning-disk confocal unit is coupled to the left port. It is equipped with two integrated laser engines (ILE) that control seven fluorescence excitation lasers and two back-illuminated sCMOS cameras, enabling for dual fluorophore imaging in parallel Abb: TI, Transilluminator; FS, field stop; AOD, acustooptical deflector; HWP, half wave plate; CAM, camera (G) Photograph of the optical trapping equipment. Red circle indicates the Bertrand lens, that can be switched into the optical path manually. Please click here to view a larger version of this figure.
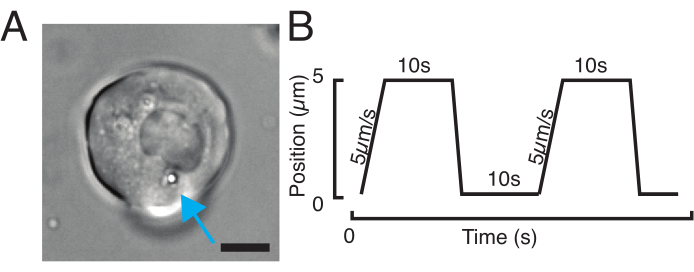
Figure 3: Choosing the right samples and parameters. (A) Representative image of an isolated zebrafish progenitor stem cell with a single microsphere positioned close enough to the nucleus to perform the indentation experiment. Scale bar = 10 µm. (B) Exemplary trap trajectory; indentation depth 5 µm; indentation speed = 5 µm/s; relaxation time 10 s. Please click here to view a larger version of this figure.
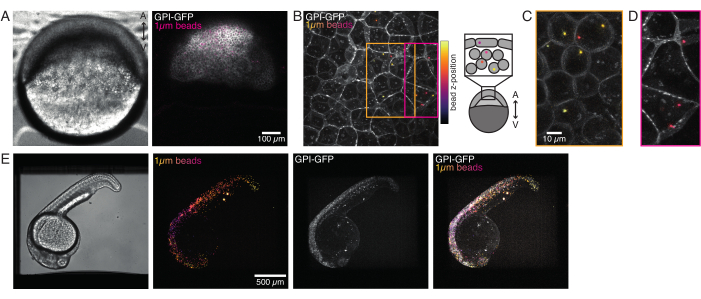
Figure 4: Microbead localization inside zebrafish embryos during development. 0.5 nL of 1 µm red fluorescent beads are injected together with GPI-GFP mRNA (100 pg/embryo, plasma membrane) in WT embryos to visualize bead localizations. (A–D) Distribution of microsphere 5 h post injection inside an embryo mounted in 0.75% agarose. (A) Brightfield and fluorescence image. The beads are homogenously dispersed across the embryo tissue as seen in a confocal micrograph. (B) Maximum projection of confocal fluorescence z-stack. The beads are color-coded from purple to yellow according to their z-position in the image stack. Purple/magenta corresponds to the most outer beads/cells (EVL; epithelial enveloping layer; or progenitor stem cells located close to the EVL surface), yellow corresponds to inner beads (progenitor deep cells), as shown in the sketch on the right. (C) Cut and maximum projection of a sub-stack of (B) corresponding to the region in the orange box: a large fraction of deep cells contain 1-2 beads. (D) Cut and maximum projection of a sub-stack of (B) corresponding to magenta box: some EVL cells contain 1-2 beads. (E) Brightfield image and maximum projection of a z-stack of a 24 hpf embryo mounted in 0.75% agarose and anesthetized with tricaine. Embryos were pre-incubated with tricaine for 15 min. From left to right: microspheres (1 µm diameter), GPI-GFP and image overlap. The beads distributed across the entire body of the embryo. Scalebar dimension indicated in each panel. Please click here to view a larger version of this figure.
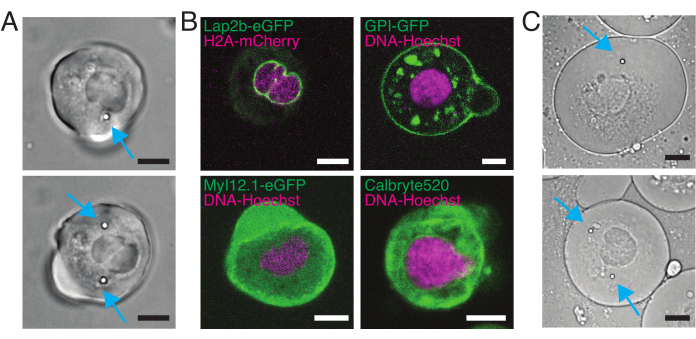
Figure 5: Isolated zebrafish progenitor stem cells with different labeling. (A) Transmission light microscopy image of suspension cells with 1 (top) or 2 (bottom) injected beads. Cyan arrows point at beads. (B) Fluorescent confocal images of suspension cells with different stainings. Top-left: Lap2b-eGFP (inner nuclear membrane, 80 pg/embryo) and H2A-mCherry. Top-right: GPI-GFP (plasma membrane, 100 pg/embryo) and DNA-Hoechst (stained as described in section 2). Bottom-left: MyI12.1-eGFP (transgenic line) and DNA-Hoechst. Bottom-right: Calbryte488 and DNA-Hoechst (stained as described in section 2). (C) Transmission light microscopy image of confined cells with 1 (top) or 2 (bottom) injected beads. Cyan arrows point at beads. Scale bars = 10 µm. Please click here to view a larger version of this figure.
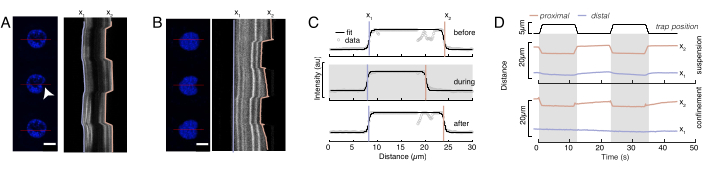
Figure 6: Estimating nuclear deformation from spinning disc movies. (A,B) Time-lapse of an indentation experiment of the nucleus in (A) a suspended cell and (B) a confined cell. Scale bar 10 µm. Representative snapshots of a Hoechst-labeled nuclei are shown 5 s before, during, and 5 s after indentation with an optically-trapped microsphere (white arrowhead). Kymographs along the indentation segment (red line, right panel). x1 and x2 are the distal and proximal (close to the bead) boundaries of the nucleus during the indentation experiment extracted from the fit of the intensity profile to Equation 1. (C) Intensity profiles along the indentation segment for three different frames (before, during and after indentation) and fitted to Equation 1 to assess the distal, x1, and proximal, x2, positions of the nucleus edges. (D) Representative trajectories of x1(t) in blue and x2(t) in amber during an indentation experiment of suspended and confined cells (10 µm). Shaded areas indicate the indentation, the distance between x1 and x2 indicates the nucleus diameter. Please click here to view a larger version of this figure.
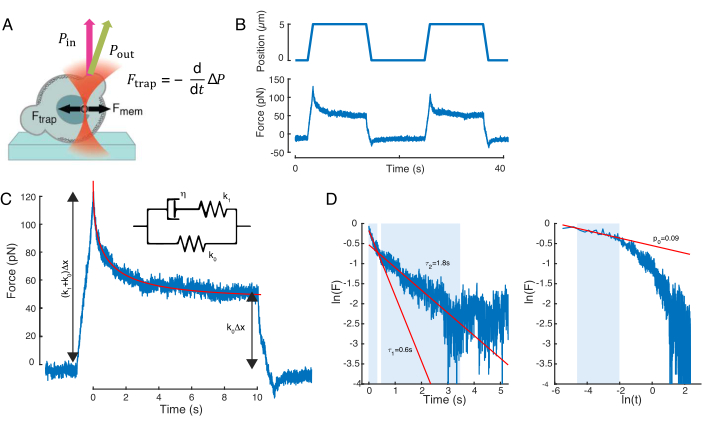
Figure 7: Force signal processing. (A) Schematic of an optically-trapped microsphere deforming the cell nucleus upon indentation. Nuclear membrane and optical forces are indicated by the black arrows. The change in beam momentum is indicated by the green arrow Pout. (B) Trap trajectory (top) and force (bottom) experienced by the optically-trapped microsphere during a repeated nuclear indentation experiment. (C) Force relaxation decay after the force peak at the maximal indentation depth. Inset shows a schematic of standard linear solid whose dynamics approximate the phenomenological observations here. (D) Left: logarithm of the normalized force versus time. The shadowed areas indicate the data portion used to fit the double exponential decay (red lines). Right: logarithm of the normalized force versus the logarithm of time. The shadowed area indicates the data portion used to fit the power law. Please click here to view a larger version of this figure.
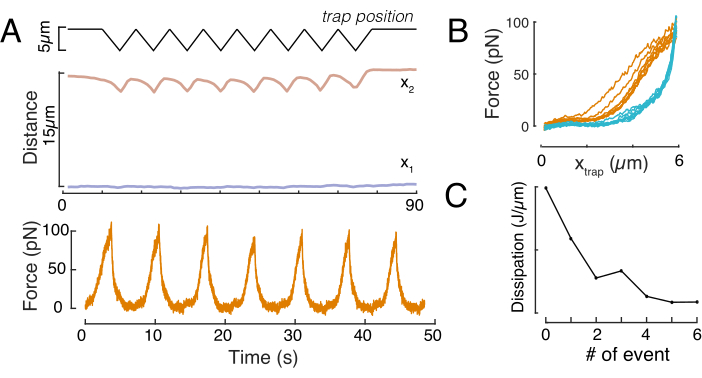
Figure 8: Force indentation routine with triangular trap displacements. (A) Representative trajectory of x1(t) in blue and x2(t) in amber during a triangular indentation experiment taken on a cell in 10 µm confinement height. Top: Trap position. Middle: Nucleus shape analysis. The distance between x1 and x2 indicates the nucleus diameter. Bottom: Force signal. (B) Force vs trap position for eight consecutive indentations. (C) Evolution of the dissipation, derived from the hysteresis between the approach and withdrawal part of the f-d curve, of the nucleus for each subsequent indentation event. Please click here to view a larger version of this figure.
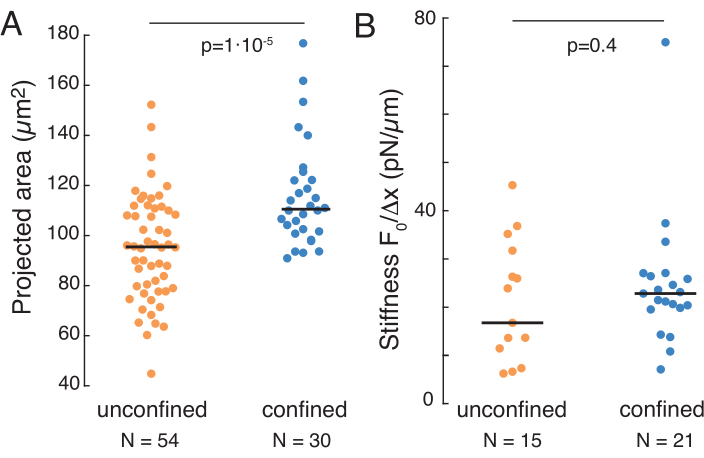
Figure 9. Nuclear properties of cells in suspension (adhesive surface) and confinement from trapezoidal routines. (A) Projected area of the nucleus from cells in suspension and under 10 µm confinement. Black bar represents the median. (B) Nuclear stiffness of cells in suspension and under confinement. Black bar represents the median. P-values derived from Kruskal-Wallis test using MatLab. Please click here to view a larger version of this figure.
Supplemental Table 1: Trapezoidal trajectory defined by the optical tweezers software. First (second) row is the x (y) distance that the trap will be linearly displaced. On the third row, the duration of a given step is set in seconds. This trajectory is composed of seven points and corresponds to the trapezoid loaded two times against the nucleus in Figure 7B. Please click here to download this Table.
Supplemental Table 2: Triangular trajectory defined by the optical tweezers software. Analogous to Table 2, this trajectory is composed of 16 points, corresponding to eight indentation events at a depth of 5 µm and a velocity of 2.5 µm/s. Please click here to download this Table.
Supplemental Table 3: Fitting parameters for the data in Figure 7. IG: initial guess. Please click here to download this Table.
Supplemental Figure S1: Optical force sensor alignment and momentum baseline compensation. (A) Field stop imaged at the auxiliary camera (AUX, Figure 2) through the Bertrand lens. An air bubble appears visible in the immersion oil, which is not visible through the eyepiece. (B) Clean optical path. For accurate alignment, open the field stop and make it coincide with the NA = 1.2 light cone. (C) Image of the sample plane. The red square indicates the OT working area. Scale bar: 20 µm. (D) Trap power measured across the FOV, along white double arrows indicated in C. In red, trap power variation when no correction is applied. In blue, trap power corrected over the entire field of view. (E) X-component of the momentum baseline along the same range. In red, non-corrected trace. In blue, trace corrected for trap power. In green, trace corrected for momentum baseline using Global Offset Compensation in the manufacturer's software. (F) Same as in E, for the Y-component. Note that under normal operation, the shaded components are used for mechanics and force measurements, e.g., x force component during movement along the x coordinate and the y force component during movement along the y-axis. After all the corrections are implemented, an RMSD noise of <0.5 pN is obtained. Please click here to download this File.
Supplemental Figure S2: A failed routine due to weak traps. (A) Kymograph showing a nucleus indentation from a failed routine. Only short, transient deformations are visible due to an escape of the bead from the trap. Importantly, the trapping laser still moves without bead to complete the predefined trajectory (green dotted line). Scale bar = 10 µm. (B) Top: Trap position versus time. Middle: Edge tracking result of the indented proximal and distal nucleus edge. Note that the distal edge is not moving without the indentation as commonly observed for completed routines on isolated cells on adhesive substrates. Bottom: Force versus time showing the loss of the microsphere indicated by a reduction in thermal noise and a sudden drop to zero force. Please click here to download this File.
Supplemental Figure S3: Survival of injected embryos. Embryos injected with 1 µm beads and 100 pg/embryo of mRNA at concentrations outlined in the protocol were compared to uninjected embryos and show no significant differences 24 h post fertilization. Mean and standard deviation of three independent experiments with N > 21 embryos per condition for each experiment. Please click here to download this File.
