Automatisierte zweidimensionale raumzeitliche Analyse mobiler Einzelmolekül-FRET-Sonden
Summary
Dieser Artikel stellt eine Methode zur raumzeitlichen Analyse von mobilen, einzelmolekularen Förster-Resonanz-Energietransfer-basierten (smFRET)-basierten Sonden mittels Weitfeldfluoreszenzmikroskopie vor. Das neu entwickelte Software-Toolkit ermöglicht die Bestimmung von smFRET-Zeitspuren bewegter Sonden, einschließlich der korrekten FRET-Effizienz und der molekularen Positionen, als Funktionen der Zeit.
Abstract
Der Einzelmolekül-Förster-Resonanz-Energietransfer (smFRET) ist eine vielseitige Technik, die Entfernungen im Sub-Nanometer- bis Nanometer-Bereich meldet. Es wurde in einer Vielzahl von biophysikalischen und molekularbiologischen Experimenten eingesetzt, einschließlich der Messung molekularer Kräfte, der Charakterisierung der Konformationsdynamik von Biomolekülen, der Beobachtung der intrazellulären Kolokalisierung von Proteinen und der Bestimmung von Rezeptor-Liganden-Interaktionszeiten. In einer Weitfeldmikroskopiekonfiguration werden Experimente typischerweise mit oberflächenimmobilisierten Sonden durchgeführt. Hier wird eine Methode vorgestellt, die Einzelmolekül-Tracking mit alternierenden Anregungsexperimenten (ALEX) smFRET-Experimenten kombiniert und die Erfassung von smFRET-Zeitspuren von oberflächengebundenen, aber mobilen Sonden in Plasmamembranen oder glasgestützten Lipiddoppelschichten ermöglicht. Für die Analyse der aufgezeichneten Daten wurde eine automatisierte Open-Source-Softwaresammlung entwickelt, die (i) die Lokalisierung von Fluoreszenzsignalen, (ii) die Verfolgung einzelner Partikel, (iii) die Bestimmung von FRET-bezogenen Größen einschließlich Korrekturfaktoren, (iv) die strenge Verifizierung von smFRET-Spuren und (v) die intuitive Darstellung der Ergebnisse unterstützt. Die generierten Daten können bequem als Input für die weitere Exploration über spezielle Software verwendet werden, z.B. zur Beurteilung des Diffusionsverhaltens von Sonden oder zur Untersuchung von FRET-Übergängen.
Introduction
Der Förster-Resonanzenergietransfer (FRET) ist ein wichtiger Treiber in der molekularbiologischen und biophysikalischen Forschung, da er die Untersuchung von Prozessen mit Sub-Nanometer-Auflösung ermöglicht. Da die Effizienz des Energietransfers zwischen Donor- und Akzeptorfluorophoren stark vom Interfarbstoffabstand im Sub-Nanometer- bis Nanometer-Bereich abhängt, wurde es effektiv als spektroskopisches Lineal zur Untersuchung der statischen und dynamischen Konformation von Biomolekülen eingesetzt1,2,3,4. Darüber hinaus wurde das FRET-Phänomen in großem Umfang für Kolokalisationsstudien von membranassoziierten und intrazellulären Proteinen auf Bulk-Ebene verwendet5,6. In den letzten zwei Jahrzehnten wurde die Methode für die Überwachung von smFRET-Ereignissen7 angepasst, was dazu beitrug, die zeitliche und räumliche Auflösung erheblich zu erhöhen und selbst seltene Subpopulationen in heterogenen Proben aufzulösen. Ausgestattet mit diesen Techniken wurden einzigartige Einblicke in die Dynamik molekularer Maschinen gewonnen, wie die Transkriptverarbeitungsrate der RNA-Polymerase II8, die Replikationsgeschwindigkeit von DNA-Polymerasen9,10, die Nukleosomen-Translokationsrate11, die Transkript-Spleiß- und Stillstandsrate von zusammengesetzten Spleißosomen12, die Aktivität von ribosomalen Subpopulationen13 und die Gehgeschwindigkeit von Kinesinmotoren14 , um nur einige zu nennen. Die Dauer der Rezeptor-Liganden-Interaktion15 und die molekularen Kräfte16 wurden quantifiziert.
Intensitätsbasierte smFRET-Studien stützen sich typischerweise auf sensibilisierte Emissionen, um die FRET-Effizienz zu messen: Ein Strahlteiler im Emissionspfad trennt bei Donoranregung räumlich Licht, das von Donor- und Akzeptorfluorophoren stammt, was die Quantifizierung einzelner Fluoreszenzintensitäten ermöglicht. Der Wirkungsgrad kann anschließend als Bruchteil der vom Akzeptor emittierten Photonen in Bezug auf die Gesamtphotonenzahl berechnet werden17. Darüber hinaus ermöglicht die Akzeptoranregung nach Donoranregung (ALEX) die Messung der Stöchiometrie der FRET-Ereignisse, was die Unterscheidung zwischen echten low FRET-Signalen von Signalen unterstützt, die z.B. von Sonden mit einem photogebleichten Akzeptor fluorophor18 auftreten.
Einzelmolekül-FRET-Experimente werden üblicherweise auf eine von zwei Arten durchgeführt. Zunächst wird ein kleiner Bereich im Probenvolumen mit einem konfokalen Mikroskop beleuchtet. Einzelne Sondenmoleküle in Lösung werden angeregt, wenn sie innerhalb des Brennvolumens diffundieren. Mit dieser Technik können schnelle Photonenzähldetektoren verwendet werden, die eine Zeitauflösung im Submikrosekundenbereich ermöglichen. Zweitens werden Sonden speziell auf Oberflächen immobilisiert und über die Weitfeldmikroskopie überwacht, wobei häufig eine TIR-Konfiguration (Total Internal Reflection) verwendet wird, um die Hintergrundfluoreszenz zu minimieren. Die Sondenimmobilisierung ermöglicht viel längere Aufnahmezeiten als mit dem ersten Ansatz. Zudem erlaubt das größere Sichtfeld die parallele Überwachung mehrerer Sonden. Die Notwendigkeit einer Kamera macht diese Methode im Vergleich zu der oben beschriebenen langsam. Die Zeitauflösung ist auf den Millisekunden- bis Sekundenbereich beschränkt.
Werden Langzeitspuren benötigt, z.B. um dynamische Prozesse auf einer Millisekunden- bis Sekundenskala zu untersuchen, ist die erste Methode nicht anwendbar, da die Fluoreszenzausbrüche typischerweise zu kurz sind. Der zweite Ansatz scheitert immer dann, wenn eine Immobilisierung nicht möglich ist, z.B. in Lebendzellexperimenten mit Sonden, die innerhalb der Zellmembran diffundieren. Darüber hinaus wurde beobachtet, dass biologische Modellsysteme ihre Reaktion in Abhängigkeit von der Mobilität der kontaktierten Oberfläche dramatisch variieren können16.
Während in der Vergangenheit kombinierte smFRET- und Einzelpartikel-Tracking-Experimente zur Aufzeichnung mobiler FRET-Sonden durchgeführt wurden19, gibt es keine öffentlich verfügbare Software für die Auswertung der Daten. Dies führte zur Entwicklung einer neuen Analyseplattform, die die Bestimmung mehrerer Eigenschaften mobiler Fluoreszenzsonden ermöglicht, einschließlich smFRET-Effizienz und Stöchiometrie, Positionen mit Subpixelgenauigkeit und Fluoreszenzintensitäten als Zeitfunktionen. Methoden zur Filterung der resultierenden Spuren durch untersuchung des schrittweisen Bleichverhaltens, der Abstände des nächsten Nachbarn, der Emissionsintensitäten und anderer Merkmale wurden etabliert, um ausschließlich korrekt synthetisierte und funktionelle Einzelsondenmoleküle auszuwählen. Die Software unterstützt auch experimentelle und analytische Techniken, die kürzlich in einer Mehrlaborstudie vereinbart wurden, um zuverlässige, quantitative smFRET-Daten zu produzieren17. Insbesondere hält sich die Implementierung an die validierten Verfahren zur Berechnung der FRET-Effizienz und Stöchiometrie. Für die Berechnung des scheinbaren FRET-Wirkungsgrades Eapp mit Eq (1) werden Fluoreszenzintensitäten bei Donoranregung im Donoremissionskanal IDD und Akzeptor-Emissionskanal IDA verwendet.
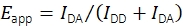 (1)
(1)
Mit Hilfe der Fluoreszenzintensität im Akzeptoremissionskanal bei Akzeptoranregung IAA wird die scheinbare Stöchiometrie mit Eq (2) berechnet.
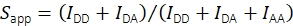 (2)
(2)
Der FRET-Wirkungsgrad E und die Stöchiometrie S können aus Eapp und Sapp unter Berücksichtigung von vier Korrekturfaktoren abgeleitet werden.
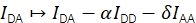
α beschreibt das Austreten der Donorfluoreszenz in den Akzeptoremissionskanal und kann unter Verwendung einer Probe, die nur Donorfluorophore enthält, oder durch Analyse von Teilen von Trajektorien, in denen der Akzeptor gebleicht wurde, bestimmt werden. δ korrigiert die direkte Anregung des Akzeptors durch die Donoranregungslichtquelle und kann mit einer Probe mit nur Akzeptorfluorophoren oder durch Analyse von Teilen von Trajektorien, in denen der Spender gebleicht wurde, gemessen werden.
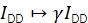 .
.
γ Skaliert IDD, um divergierende Detektionseffizienzen in Donor- und Akzeptoremissionskanälen und unterschiedliche Quantenwirkungsgrade der Fluorophore zu korrigieren. Der Faktor kann berechnet werden, indem die Zunahme der Donorintensität beim Akzeptorbleichen in Trajektorien mit hoher FRET-Effizienz20 analysiert wird oder indem eine Probe mit mehreren diskreten FRET-Zuständen untersucht wird.
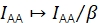
β skaliert IAA, um unterschiedliche Wirkungsgrade der Donor- und Akzeptorerregung zu korrigieren. Wenn γ mittels Akzeptorbleichanalyse bestimmt würde, könnte β aus einer Stichprobe des bekannten Donor-zu-Akzeptor-Verhältnisses berechnet werden21. Andernfalls liefert die Mehrzustands-FRET-Probe auch β.
Zusammen ermöglichen die Korrekturen die Berechnung des korrigierten FRET-Wirkungsgrades mit Eq (3).
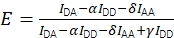 (3)
(3)
und die korrigierte Stöchiometrie mit Eq (4).
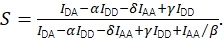 (4)
(4)
Idealerweise ergibt die korrigierte Stöchiometrie für ein Donor-zu-Akzeptor-Verhältnis von 1:1 S = 0,5. In der Praxis erzeugt ein reduziertes Signal-Rausch-Verhältnis eine Streuung der Messwerte von S, was die Unterscheidung von reinen Donorsignalen (S = 1) und reinen Akzeptorsignalen (S = 0) behindert. Die resultierenden Zeitspuren können als Eingabe für eine detailliertere Analyse der Einzelmolekültrajektorien verwendet werden, um Informationen wie raumzeitliche Kraftprofile16, die Mobilität der Einzelmolekülereignisse22 oder die Übergangskinetik zwischen verschiedenen Zuständen1 zu erhalten.
Das folgende Protokoll beschreibt experimentelle Parameter und Verfahren für smFRET-Tracking-Experimente sowie das Arbeitsprinzip der Datenanalyse mit der neu entwickelten Software-Suite. Für die Erfassung experimenteller Daten wird empfohlen, einen Mikroskopieaufbau zu verwenden, der die folgenden Anforderungen erfüllt: i) Fähigkeit, die Emission einzelner Farbstoffmoleküle nachzuweisen; ii) Weitfeldbeleuchtung: Insbesondere für Experimente mit lebenden Zellen wird die Konfiguration der inneren Totalreflexion (TIR23,24,25) empfohlen; iii) räumliche Trennung des Emissionslichts nach Wellenlänge, so dass die Donor- und Akzeptorfluoreszenz auf verschiedene Bereiche desselben Kamerachips25 oder verschiedener Kameras projiziert wird; iv) Modulation von Lichtquellen zur Donor- und Akzeptoranregung mit Millisekundengenauigkeit, z.B. mit direkt modulierbaren Lasern oder Modulation über akusto-optische Modulatoren. Dies ermöglicht eine stroboskopische Beleuchtung, um das Photobleichen von Fluorophoren sowie die abwechselnde Anregung zur Bestimmung von Stöchiometrien zu minimieren. v) Ausgabe einer Datei pro aufgezeichneter Bildsequenz in einem Format, das vom PIMS-Python-Paket gelesen werden kann26. Insbesondere werden mehrseitige TIFF-Dateien unterstützt.
Protocol
Representative Results
Discussion
Dieser Artikel beschreibt eine Pipeline für die automatisierte Aufzeichnung und quantitative Analyse von smFRET-Daten, die von mobilen, aber oberflächengebundenen Sondenmolekülen stammen. Es ergänzt die beiden vorherrschenden Ansätze für smFRET-Experimente, an denen entweder oberflächenimmobilisierte Sonden oder sonden beteiligt sind, die in Lösung in ein konfokales Anregungsvolumen hinein und aus ihm heraus diffundieren17. Es liefert die richtige FRET-Effizienz und die molekularen Positionen als Funktion der Zeit. Es kann daher als Input für spezialisierte Analyseprogramme verwendet werden, z. B. zur Quantifizierung der Übergangskinetik1, der FRET-Histogramme39 oder der zweidimensionalen Diffusion22.
Die Software wird unter einer freien und Open-Source-Lizenz veröffentlicht, die von der Open Source Initiative genehmigt wurde und dem Benutzer das unbefristete Recht auf freie Nutzung, Änderung und Weiterverbreitung gewährt. Github wurde als Entwicklungs- und Distributionsplattform ausgewählt, um es so einfach wie möglich zu machen, die Software zu erhalten und am Entwicklungsprozess teilzunehmen, indem Fehler gemeldet oder Code40 beigetragen wird. In Python geschrieben, ist die Software nicht von proprietären Komponenten abhängig. Die Wahl der Jupyter Notebooks als Benutzeroberfläche erleichtert die Überprüfung der Daten bei jedem Analyseschritt und ermöglicht es, die Pipeline speziell auf das vorliegende Versuchssystem zuzuschneiden und zu erweitern. Die sdt-python library32 dient als Grundlage und implementiert Funktionen zur Auswertung von Fluoreszenzmikroskopiedaten, wie z. B. Einzelmoleküllokalisation, Diffusionsanalyse, Fluoreszenzintensitätsanalyse, Farbkanalregistrierung, Kolokalisierungsanalyse und ROI-Handling.
Prinzipiell kann die Einzelpartikelverfolgung in ein-, zwei- oder dreidimensionalen Systemen durchgeführt werden. Hier wurde die Einzelmolekül-Analyse-Pipeline auf die Untersuchung mobiler 2D-Systeme zugeschnitten. Diese Wahl spiegelt die Verfügbarkeit einfacher Systeme wie planar unterstützter Lipiddoppelschichten (SLBs) wider, um mobile Fluoreszenzsonden zu präsentieren. Solche Lipiddoppelschichtsysteme bestehen typischerweise aus zwei oder mehr Phospholipid-Einheiten, wobei die Bulk-Fraktion die wichtigsten physikalisch-chemischen Parameter des SLB (wie Phase und Viskosität) bestimmt und die Minor-Fraktion Anheftungsstellen für Biomoleküle bereitstellt. Diese Bindungsstellen können biotinylierte Phospholipide für Avidin- oder Streptavidin-basierte Proteinplattformen oder Nickel-NTA-konjugierte Phospholipide für Proteinplattformen mit Histidin-Tags41 sein. Die Wahl der geeigneten Plattform zur Verknüpfung von Proteinen mit dem SLB hängt von der wissenschaftlichen Fragestellung ab. Leser können sich auf die Literatur16,38,42 für Beispiele für erfolgreich eingesetzte Strategien beziehen. Die Dichte der Sonden in der Probe sollte ausreichend niedrig sein, um überlappende Punktspreizungsfunktionen zu vermeiden. Typischerweise werden weniger als 0,1 Moleküle pro μm2 empfohlen. Ein Beispiel für eine geeignete Sondendichte finden Sie im Abschnitt repräsentative Ergebnisse (insbesondere Abbildung 6). Die Analysemethode ist auch auf einzelne fluoreszenzmarkierte Proteinmoleküle anwendbar, die in der Plasmamembran lebender Zellen diffundieren.
Ein kritischer Aspekt der smFRET-Experimente ist die Herstellung und Charakterisierung der FRET-Sonden selbst. Bei der Auswahl von Fluorophoren für ein FRET-Paar sollte ihr Försterradius mit den erwarteten Interfarbstoffabständen übereinstimmen43. Farbstoffe, die gegen Photobleichen beständig sind, werden bevorzugt, da sie langzeitige Spuren ergeben. Für erhöhte Bleichraten kann jedoch eine Fluorophorspezies verwendet werden, um Multiemitter-Ereignisse zu erkennen, die von kolokalisierten Molekülen über eine schrittweise Photobleichanalyse ausgehen; Siehe Schritt 8.1.4 im Abschnitt Protokoll. Fluorophorpaare sollten ortsspezifisch und kovalent an die interessierenden Moleküle gebunden sein und intra- oder intermolekulare FRET-Paare bilden.
Die Kombination von smFRET mit anderen leicht verfügbaren Techniken kann seine räumliche Auflösung über die Beugungsgrenze hinaus erhöhen (über STED44). Der hier vorgestellte smFRET-Tracking-Algorithmus erweitert die Anwendbarkeit des Ansatzes auf neue experimentelle Settings und Modellsysteme. Dazu gehören Untersuchungen (i) kinetischer Veränderungen in der Stöchiometrie mobiler Biomoleküle, (ii) der dynamischen Assoziation mobiler Biomoleküle, (iii) der Geschwindigkeit enzymatischer Reaktionen frei diffundierender Reaktanten und (iv) der Kinetik von Konformationsänderungen mobiler Biomoleküle. Die ersten beiden Beispiele erfordern Modellsysteme, die eine intermolekulare FRET zeigen, d.h. Donor und Akzeptor werden zu getrennten biomolekularen Entitäten von Interesse konjugiert. Die letzteren Beispiele können Biosensoren verwenden, die Donor und Akzeptor innerhalb derselben molekularen Einheit (intramolekulare FRET) tragen.
Intramolekulare FRET-basierte Sensoren können Einblicke in intrinsische Konformationsänderungen von Biomolekülen1,2,3,4, Konformationsänderungen durch endogene oder externe Kraftbelastung (molekulare Kraftsensoren16) oder Ionenkonzentrationen in der Nanoumgebung wie calcium45 und pH46 geben . Abhängig vom Modellsystem und der bevorzugten Verankerungsplattform können solche smFRET-Ereignisse entweder in 2D oder 3D verfolgt werden: (i) Planares Tracking von smFRET-Ereignissen kann zur Quantifizierung von Rezeptor-Liganden-Interaktionszeiten innerhalb einer Plasmamembran, der Assoziation von membranverankerten Signalverstärkungskaskaden und den stöchiometrischen Veränderungen von Oberflächenrezeptoren eingesetzt werden; ii) Die Volumenverfolgung von smFRET-Ereignissen kann für alle intra- oder intermolekularen FRET-Sonden in lebenden Zellen oder in in vitro rekonstituierten Systemen verwendet werden.
Die smFRET-Tracking-Methode wurde hauptsächlich mit Blick auf intramolekulare FRET-Sonden entwickelt. Diese Sonden weisen eine feste und bekannte Anzahl von fluoreszierenden Markierungen auf, eine Tatsache, die ausgenutzt wurde, um Daten von agglomerierten und falsch synthetisierten (z. B. unvollständig markierten) Molekülen sowie von Sonden, bei denen eines der Fluorophore photogebleicht wurde, abzulehnen. Durch die Einstellung der Filterschritte kann das Verfahren jedoch auch auf intermolekulare FRET-Sonden angewendet werden. Zum Beispiel könnte man, anstatt nur Moleküle mit einem einzelnen Donor- und einem einzelnen Akzeptorfluorophor zu akzeptieren, die räumlichen Trajektorien von Donor- und Akzeptorfarbstoffen untersuchen und beispielsweise für die Co-Diffusion von Donor-Akzeptor-Trajektorien auswählen.
Da der 3D-DAOSTORM-Algorithmus die Bestimmung der Position eines Signals entlang der optischen Achse über den Astigmatismus aufgrund einer zylindrischen Linse im Emissionsstrahlpfad unterstützt, könnten 3D-Experimente problemlos in die Analysepipeline integriert werden. In diesem Fall würde das Akzeptorsignal bei Akzeptoranregung dazu dienen, die Stöchiometrie und die axiale Position zu bestimmen. Die Analysesoftware kann auch verwendet werden, um Daten aus Experimenten mit immobilisierten Sonden auszuwerten, indem sie ihren hohen Automatisierungsgrad und filternde Schemata nutzt. Tatsächlich wurden smFRET-Effizienzdatensätze von Holliday-Übergängen, die auf Gelphasendoppelschichten immobilisiert wurden38 , mit einer frühen Version der Software analysiert.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Unterstützt wurde diese Arbeit durch die Projekte P30214-N36, P32307-B des Wissenschaftsfonds (FWF) und durch den Vienna Science and Technology Fund (WWTF) LS13-030.
Materials
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt) (Ni-NTA-DOGS) | Avanti Polar Lipids | 790404P | |
| 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) | Avanti Polar Lipids | 850375P | |
| 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | 850457P | |
| α Plan-FLUAR 100x/1.45 oil objective | Zeiss | 000000-1084-514 | |
| Axio Observer microscope body | Zeiss | ||
| Bandpass filter | Chroma Technology Corp | ET570/60m | donor emission filter |
| Bandpass filter | Chroma Technology Corp | ET675/50m | acceptor emission filter |
| conda-forge | conda-forge community | community-maintaned Python package repository for Anaconda/miniconda | |
| Coverslips 60 mm x 24 mm #1.5 | MENZEL | ||
| Dichroic mirror | Semrock Inc | FF640-FDi01-25×36 | separation of donor and acceptor emission |
| Dichroic mirror (quad band) | Semrock Inc | Di01-R405/488/532/635-25×36 | separation of excitation and emission light |
| DPBS | Sigma-Aldrich | D8537 | |
| FCS | Sigma-Aldrich | F7524 | for imaging buffer |
| fret-analysis | Schütz group at TU Wien | Python package for smFRET data analysis; version 3 | |
| Fura-2 AM | Thermo Fisher Scientific | 11524766 | |
| HBSS | Sigma-Aldrich | H8264 | for imaging buffer |
| iBeam Smart 405-S 405 nm laser | Toptica Photonics AG | ||
| iXon Ultra 897 EMCCD camera | Andor Technology Ltd | ||
| Lab-Tek chambers (8 wells) | Thermo Fisher Scientific | 177402PK | for sample preparation and imaging |
| Millenia Prime 532 nm laser | Spectra Physics | ||
| miniconda | Anaconda Inc. | Python 3 distribution. Min. version: 3.7 | |
| Monovalent streptavidin (plasmids for bacterial expression) | Addgene | 20860 & 20859 | |
| OBIS 640 nm laser | Coherent Inc | 1185055 | |
| Optosplit II | Cairn Research | ||
| Ovalbumin | Sigma-Aldrich | A5253 | for imaging buffer |
| Plasma cleaner | Harrick Plasma | PDC-002 | |
| sdt-python | Schütz group at TU Wien | Python library for data analysis; version 17 | |
| TetraSpek bead size kit | Thermo Fisher Scientific | T14792 | Randomly distributed, immobilized fiducial markers for image registration |
| USC500TH Ultrasound bath | VWR | for SUV formation |
References
- McKinney, S. A., Déclais, A. -. C., Lilley, D. M. J., Ha, T. Structural dynamics of individual holliday junctions. Nature Structural Biology. 10 (2), 93-97 (2002).
- Wang, S., Vafabakhsh, R., Borschel, W. F., Ha, T., Nichols, C. G. Structural dynamics of potassium-channel gating revealed by single-molecule FRET. Nature Structural & Molecular Biology. 23 (1), 31-36 (2015).
- Hellenkamp, B., Wortmann, P., Kandzia, F., Zacharias, M., Hugel, T. Multidomain structure and correlated dynamics determined by self-consistent FRET networks. Nature Methods. 14 (2), 174-180 (2016).
- Kilic, S., et al. Single-molecule FRET reveals multiscale chromatin dynamics modulated by HP1α. Nature Communications. 9 (1), 235 (2018).
- Stryer, L. Fluorescence energy transfer as a spectroscopic ruler. Annual Review of Biochemistry. 47 (1), 819-846 (1978).
- Wu, P. G., Brand, L. Resonance energy transfer: Methods and applications. Analytical Biochemistry. 218 (1), 1-13 (1994).
- Qiao, Y., Luo, Y., Long, N., Xing, Y., Tu, J. Single-molecular förster resonance energy transfer measurement on structures and interactions of biomolecules. Micromachines. 12 (5), 492 (2021).
- Malkusch, N., Dörfler, T., Nagy, J., Eilert, T., Michaelis, J. smFRET experiments of the RNA polymerase II transcription initiation complex. Methods. 120, 115-124 (2017).
- Lee, J. -. B., et al. Single-molecule views of MutS on mismatched DNA. DNA repair. 20, 82-93 (2014).
- Phelps, C., Israels, B., Jose, D., Marsh, M. C., von Hippel, P. H., Marcus, A. H. Using microsecond single-molecule FRET to determine the assembly pathways of T4 ssDNA binding protein onto model DNA replication forks. Proceedings of the National Academy of Sciences of the United States of America. 114 (18), E3612-E3621 (2017).
- Deindl, S., Zhuang, X. Monitoring conformational dynamics with single-molecule fluorescence energy transfer: Applications in nucleosome remodeling. Methods in Enzymology. 513, 59-86 (2012).
- Crawford, D. J., Hoskins, A. A., Friedman, L. J., Gelles, J., Moore, M. J. Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. Proceedings of the National Academy of Sciences of the United States of America. 110 (17), 6783-6788 (2013).
- Wang, Y., Xiao, M., Li, Y. Heterogeneity of single molecule FRET signals reveals multiple active ribosome subpopulations. Proteins. 82 (1), 1-9 (2014).
- Mori, T., Vale, R. D., Tomishige, M. How kinesin waits between steps. Nature. 450 (7170), 750-754 (2007).
- Huppa, J. B., et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 463 (7283), 963-967 (2010).
- Göhring, J., et al. Temporal analysis of T-cell receptor-imposed forces via quantitative single molecule FRET measurements. Nature Communications. 12 (1), 2502 (2021).
- Hellenkamp, B., et al. Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nature Methods. 15 (9), 669 (2018).
- Kapanidis, A. N., et al. Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proceedings of the National Academy of Sciences of the United States of America. 101 (24), 8936-8941 (2004).
- Sakon, J. J., Weninger, K. R. Detecting the conformation of individual proteins in live cells. Nature Methods. 7 (3), 203-205 (2010).
- McCann, J. J., Choi, U. B., Zheng, L., Weninger, K., Bowen, M. E. Optimizing methods to recover absolute FRET efficiency from immobilized single molecules. Biophysical Journal. 99 (3), 961-970 (2010).
- Lee, N. K., et al. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophysical Journal. 88 (4), 2939-2953 (2005).
- Asher, W. B., et al. Single-molecule FRET imaging of GPCR dimers in living cells. Nature Methods. 18 (4), 397-405 (2021).
- Joo, C., Ha, T. . Single-molecule FRET with total internal reflection microscopy. (12), 1223-1237 (2012).
- Joo, C., Ha, T. Objective-type total internal reflection microscopy (excitation) for single-molecule FRET. Cold Spring Harbor Protocols. 11, 1189-1191 (2012).
- Joo, C., Ha, T. Objective-type total internal reflection microscopy (emission) for single-molecule FRET. Cold Spring Harbor Protocols. 11, 1192-1194 (2012).
- Allan, D. B., Caswell, T., van der Wel, C. M., Dimiduk, T. . Soft-matter/pims: PIMS v0.5. , (2020).
- Anaconda Inc. . Miniconda. , (2021).
- conda-forge community. . The conda-forge project: community-based software distribution built on the conda package format and ecosystem. , (2015).
- . . JupyterLab Contributors Notebooks – JupyterLab documentation. , (2021).
- Babcock, H., Sigal, Y. M., Zhuang, X. A high-density 3D localization algorithm for stochastic optical reconstruction microscopy. Optical Nanoscopy. 1 (6), (2012).
- Gao, Y., Kilfoil, M. L. Accurate detection and complete tracking of large populations of features in three dimensions. Optics Express. 17 (6), 4685 (2009).
- Schrangl, L. . sdt-python: Python library for fluorescence microscopy data analysis (v17.1). , (2021).
- Crocker, J. C., Grier, D. G. Methods of digital video microscopy for colloidal studies. Journal of Colloid and Interface Science. 179 (1), 298-310 (1996).
- Preibisch, S., Saalfeld, S., Schindelin, J., Tomancak, P. Software for bead-based registration of selective plane illumination microscopy data. Nature Methods. 7 (6), 418-419 (2010).
- Bradski, G. The OpenCV library. Dr. Dobb’s Journal: Software Tools for the Professional Programmer. 25 (11), 120-123 (2000).
- Allan, D. B., Caswell, T., Keim, N. C., van der Wel, C. M., Verweij, R. W. Soft-matter/trackpy: Trackpy v0.5.0. Zenodo. , 4682814 (2021).
- Killick, R., Fearnhead, P., Eckley, I. A. Optimal detection of changepoints with a linear computational cost. Journal of the American Statistical Association. 107 (500), 1590-1598 (2012).
- Schrangl, L., Göhring, J., Schütz, G. J. Kinetic analysis of single molecule FRET transitions without trajectories. The Journal of Chemical Physics. 148 (12), 123328 (2018).
- Santoso, Y., Torella, J. P., Kapanidis, A. N. Characterizing single-molecule FRET dynamics with probability distribution analysis. ChemPhysChem. 11 (10), 2209-2219 (2010).
- Schrangl, L. Single-molecule FRET analysis software (3.0). Zenodo. , (2021).
- Nye, J. A., Groves, J. T. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 24 (8), 4145-4149 (2008).
- Platzer, R., et al. Unscrambling fluorophore blinking for comprehensive cluster detection via photoactivated localization microscopy. Nature Communications. 11 (1), 4993 (2020).
- Johnson, I., Spence, M. . The molecular probes handbook: A guide to fluorescent probes and labeling technologies. , (2010).
- Szalai, A. M., et al. Super-resolution imaging of energy transfer by intensity-based STED-FRET. Nano Letters. 21 (5), 2296-2303 (2021).
- Miyawaki, A., et al. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388 (6645), 882-887 (1997).
- Zhai, B., Zhai, S., Hao, R., Xu, J., Liu, Z. A FRET-based two-photon probe for in vivo tracking of pH during a traumatic brain injury process. New Journal of Chemistry. 43 (43), 17018-17022 (2019).
