NOTE: An overview of the protocol is provided in Figure 1. The parameters used for the experiments described in the present protocol can be found in Supplemental Table S1 and Supplemental Table S2.
1. Preparation of the sample solution
NOTE: The protocol is described using an arabinoxylan pentasaccharide (23-α-L-arabinofuranosyl-xylotetraose or XA2XX; see the Table of Materials) as an example.
- Preparation of the solvent: 500 µM LiCl in 50:50 H2O:MeOH (vol./vol.).
- Prepare a 100 mM stock solution of lithium chloride (LiCl) in H2O by weighing 212 mg of LiCl and add 50 mL of high-purity deionized water (H2O) in a 50 mL polypropylene conical tube. Shake until completely dissolved.
NOTE: The solvent is doped with a lithium salt to promote the formation of [M+Li]+ adducts in the ion source of the spectrometer, as it usually yields better-quality fragmentation spectra compared to other alkali adducts. The use of LiCl is recommended because organic acids (and thus their salts) have previously been found to impact IMS profiles23. - In a glass bottle, dilute the LiCl stock solution 200x: to 250 µL of the stock solution, add 24.75 mL of H2O. Add 25 mL of methanol (MeOH) to reach a final concentration of LiCl of 500 µM in 50:50 H2O:MeOH (v/v). Sonicate for 2 min to degas the solvent.
NOTE: MeOH presents a health hazard (H225, H301, H311, H331, H370); manipulate under an extractor hood wearing a lab coat, gloves, and eye protection. A proportion of 50:50 MeOH/H2O (v/v) appears to be the best solvent for the ionization of oligosaccharides; however, MeOH can be substituted by acetonitrile (ACN) if needed.
- Prepare a 100 mM stock solution of lithium chloride (LiCl) in H2O by weighing 212 mg of LiCl and add 50 mL of high-purity deionized water (H2O) in a 50 mL polypropylene conical tube. Shake until completely dissolved.
- In a 1.5 mL polypropylene tube, weigh 1 mg of the carbohydrate. Dissolve with an appropriate volume of 500 µM LiCl to reach a concentration of 1 mg/mL. Dilute to a final concentration of 10 µg/mL in 50:50 MeOH/H2O + 500 µM LiCl. Store at 4 °C.
NOTE: The concentration of 10 µg/mL was chosen to optimize the signal over all fragment ions during IMS/IMS-MS (this is for a pure compound; increase the concentration when working on mixtures). For the acquisition of reference IMS/IMS spectra, do not dilute the sample further: saturation of the MS detector prior to fragmentation is expected, although the instrument offers options to correct it (see step 3.2.).
2. Tuning of the Cyclic IMS mass spectrometer
NOTE: Software-related instructions (windows, menus, and commands) are highlighted in bold.
- Open the instrument console from the instrument control software (MS tune page, see the software details in the Table of Materials), and put the instrument in Operate mode. Wait for at least 3 h for the high voltages to stabilize in the IMS cell.
NOTE: For the best reproducibility, the voltages in the IMS cell need to be completely stabilized. Turn on the high voltages and let the instrument stabilize overnight before any cyclic IMS analysis. Furthermore, the pressure and temperature in the ion mobility cell must be kept as constant as possible. Although a readback for the pressure is available in the Vacuum tab, no readback is available for the temperature. Keep the instrument in a thermostated laboratory. The instrument used in this work operates at 1.75 mbar in a laboratory thermostated at 20 °C. - Cyclic IMS instrument setup
NOTE: Standard solutions must be infused using the built-in fluidics system for the instrument setup.- Place the fluidics containers filled with the appropriate manufacturer-provided standards on the fluidics system: Reservoir B ('Lockmass'): 10 pg/µL leucine enkephaline (LEU ENK) in 50:50 ACN/H2O + 0.1% formic acid; Reservoir C ('Calibrant'): MajorMix.
NOTE: In this protocol, the MajorMix calibration solution will be used to calibrate both the m/z and CCS dimensions. For practical reasons, an external CCS calibration will be performed (see step 5 of the protocol); hence, it is also possible to use an in-house calibrant mixture for the CCS and another calibrant for the m/z (e.g., sodium formate or sodium iodide). - On the Tune page of the Quartz console, go to the Fluidics tab. Set the sample fluidics to reservoir C and the reference fluidics to reservoir B. Infuse both solutions consecutively in the ion source to check the MS signal.
- Perform the ADC setup, detector setup (using LEU ENK), and mass calibration (see the Table of Materials for the calibration solution) from the Instrument Setup page according to the manufacturer's instructions.
- Place the fluidics containers filled with the appropriate manufacturer-provided standards on the fluidics system: Reservoir B ('Lockmass'): 10 pg/µL leucine enkephaline (LEU ENK) in 50:50 ACN/H2O + 0.1% formic acid; Reservoir C ('Calibrant'): MajorMix.
- Record an IMS acquisition of the calibration solution with a single pass separation (use this for external IMS calibration).
NOTE: The ion source and the traveling wave (TW) parameters (static wave height and wave velocity) must be kept constant during all the acquisitions (calibration and acquisitions). If the user does not have prior knowledge of the optimal parameters for his sample, this step can be performed after step 3 of the protocol (for [M+Li]+ adducts of neutral oligosaccharides, the representative results use a TW height of 16 V and TW velocity of 350 m/s, which give the best results).- From the Fluidics tab, select baffle position Sample and infuse the calibrant (see the Table of Materials) in the ion source (using the built-in fluidics system) through the 'Sample' probe at a flow rate of 10 µL/min.
- Set up a single-pass IMS sequence. From the Tune page, put the instrument in Mobility mode, and open the Cyclic Sequence Control window. Select Advanced mode. From the Cyclic Functions tab of this new window, select Add Bundle, then Single/Multipass. Wait for a sequence of mobility events to appear in the Sequence tab of the same window.
NOTE: To activate the real-time display, the user must apply the instrument parameters: click on Tune in TOF mode or Run in Mobility mode. Before switching the instrument between TOF and Mobility modes, it is necessary to Abort any running acquisition (including the Tune page display). The relative abundance of ions may vary between TOF mode and Mobility mode because of changes in the ion transmission parameters. - Adapt the sequence so that all the calibrant ions make a single pass around the cyclic IMS racetrack. Do not change the Inject time or the Eject and acquire time; however, lower the Separate time to 1 ms (in the Sequence tab). If some ions of the calibration mixture do not fit in the displayed arrival time window, change the synchronization of the IMS with the pusher of the orthogonal acceleration TOF analyzer by increasing the number of Pushes Per Bin in the ADC Settings tab.
NOTE: The times in the control sequence only control the multifunction array for ion gating. As long as the ions are engaged in their first (or nth) pass around the racetrack, they will finish said pass even if the direction of the TW has changed in the array in the meantime. Lowering the separation time to 1 ms means that the array will switch to ejection mode after 1 ms. This ensures that the faster ions will not have enough time to pass through the array and engage in a second pass before the slower ions finish their first pass. Therefore, all ions will be subjected to the same number of passes (i.e., one pass), which is necessary to perform IMS calibration. - Record a 2-min acquisition. In the Cyclic Sequence Control window, click on Acquire to open the Acquisition Settings popup window. Input the Filename, Description, and Length of Acquisition (mins) and click Save.
- Record another 2 min acquisition of the calibration solution under the same conditions as step 2.3 (use this to check the quality of the CCS calibration). In the Cyclic Sequence Control window, click on Acquire to open the Acquisition Settings popup window. Input the Filename, Description, and Length of Acquisition (mins) and click Save.
- Thoroughly wash the fluidics system with 50:50 H2O/ACN to avoid crystallization of the calibrant in the peek tubing.
3. IMS/IMS-MS acquisition
- Using a syringe pump, infuse the (lithium-doped) sample at 10 µg/mL through the sample probe at a flow rate of 10 µL/min.
- Switch the instrument to TOF mode (from the MS tune page) to check the stability of the signal. Record a full MS acquisition (1 min) of the sample, which will be useful to check the isotopic pattern and the presence of potential contaminants.
NOTE: Because the sample concentration is chosen to obtain a good ion signal for the fragments, a TOF saturation may be observed at this step. TOF saturation can be identified using the following artifacts: (i) an artificially increased MS resolution, (ii) a change in isotopic ratios, and (iii) a multitude of low-abundance peaks in-between isotopes. Use the DRE Lens (Dynamic Range enhancement, Quad/MS Profile/DRE tab of the main Tune page) to attenuate the transmission of ions and discard the saturation in TOF mode (Figure 2A,B). - Put the instrument in MSMS mode (Quad/MS Profile tab of the main Tune page) and select the mass of the targeted ion in the MSMS Mass field for isolation in the quadrupole (in the example: m/z of 685.2, corresponding to the [M+Li]+ ionic species of the arabinoxylan pentasaccharide). Record a 1 min acquisition to check the precursor isolation when processing the data.
NOTE: Lithium adducts have an isotope at -1 Da of the monoisotopic peak, which needs to be removed from the MS/MS selection window so that it will not interfere with the processing steps. It can be removed by narrowing the selection range using the LM Resolution and HM Resolution parameters in the Quad/MS Profile tab (Figure 2C). - Set up a "slicing" IMS sequence to perform a mobility-based selection of the isomer of interest.
- Switch the instrument to Mobility mode (see step 2.3.2). In the Cyclic Sequence Control window, from the Cyclic Functions Tab, select Add bundle and then Slicing. Wait for a complex sequence of mobility events to appear in the Sequence tab (Figure 3).
NOTE: It is possible to visualize each step of the IMS/IMS process: click on the Eject and Acquire event in the Sequence tab. Once highlighted in red, move it to the appropriate position within the sequence using the Up and Down buttons. - Position the Eject and Acquire event right after the first Separate event (i.e., move it at row 3 instead of row 8 in the sequence as displayed in Figure 3) and then click Run. Look for the results of the initial separation to be displayed in real time. Increase the duration of the first Separate event for a multipass separation by changing the time value for this event in the sequence until the resolution of the IMS peaks is satisfactory. Record a 1 min acquisition for reference.
NOTE: Take note of the ADC Start Delay value in the ADC Setup tab: it will be useful to check the quality of the isolation. - Click Pause. Note that the results of the initial separation are displayed, although modifications in the control sequence will not be applied until the user clicks Run again. Position the Eject and Acquire event below the Eject, Eject to Pre-Store, and Hold and Eject events. Adjust the duration of the events so that the targeted peak is in the Eject to Pre-Store region, and any other ion is either in the Eject or Hold and Eject region.
NOTE: The duration of these three events compared to the arrival time distributions (ATDs) can be visualized using the color-coded bar below the mobility spectrum in the Mobilogram tab (Figure 3). - Position the Eject and Acquire event at the end of the sequence, below the Reinject from Pre-Store and the second Separate events. Click Run to display the selected population.
NOTE: Because the selected population has left the IMS cell, all previous separation has been lost, and it is back to a single-pass separation (which is desired). - Check the quality of the isolation. To verify that only the peak of interest has been selected, perform the same separation after reinjection as before reinjection (i.e., same Separate time) as shown in Figure 4. Record a 1 min acquisition for reference.
NOTE: The users are encouraged to check the ejected population; the Eject to Pre-Store time window should be baseline level (Figure 4B). To check this, put the ADC Start Delay in Manual mode in the ADC Settings tab, and enter the delay time noted in step 3.4.2. Record a 1 min acquisition for reference. - In the Sequence tab, in the column next to the user-defined event times (the Time Abs column, highlighted in red), look for the summed times of all events. Take note of the Time Abs found on the line of the Reinject from Pre-Store event for performing the CCS calibration.
- Switch the instrument to Mobility mode (see step 2.3.2). In the Cyclic Sequence Control window, from the Cyclic Functions Tab, select Add bundle and then Slicing. Wait for a complex sequence of mobility events to appear in the Sequence tab (Figure 3).
- Fragment the targeted peak between the two rounds of IMS. Change the voltages of the reinjection step to increase the kinetic energy of the ions, and fragment them upon collision with the ion mobility gas.
- Set the duration of the Separate event directly preceding the Eject and Acquire to 1 ms (see explanation in step 2.3.3).
- On the Reinject from Pre-Store line, tick the Enable Activation box, and optimize the fragmentation with the built-in control. If the spectrum is satisfactory (e.g., the base peak is a fragment), proceed directly to step 3.5.4.
NOTE: When enabling activation, three voltages on the line will turn grey: these are the voltages that the user needs to change if manual optimization of the voltages is required (see the next step). These three voltages (Pre-Array Gradient, Pre-Array Bias, and Array Offset) form the gradient used to activate the ions. The kinetic energy of the ions will increase with the slope between the Pre-Array Bias and Array Offset (see Figure 5). The default values of the Gradient → Bias → Offset values are: without activation 85 → 70 → 45 V; maximum activation of the built-in function 185 → 170 → -5 V (+150 V). After fragmentation, do not forget to readjust the ion transmission using the DRE lens (decrease the attenuation of the signal) (see step 3.2.). - If the fragmentation is not satisfactory with the built-in control, uncheck the Enable Activation box and proceed to manually optimize the reinjection voltages. Increase the Pre-Array Gradient voltage (the Pre-Array Bias voltage must always be kept 15 V below the Pre-Array Gradient), and lower the Array Offset voltage (which can be set as negative) until the results are satisfactory.
NOTE: When manually tuning the voltages of the multifunction array, the user can switch from the 'Mobilogram' view to interactive schematics of the voltages applied in the multifunction array (PE diagram) to better visualize the voltage settings (Figure 5A). - Record a 2 min acquisition. In the acquisition pop-up window, tick the Retain Drift Time option to generate a file containing only the arrival times vs m/z (the acquisition time used for chromatographic analyses-the retention time-is removed from the file). Note that this file is labeled *_dt.RAW.
NOTE: If the user forgets to check the Retain Drift Time option, it is still possible to extract the IMS dimension using the Driftscope 2.9 software (File | Export to MassLynx | Retain Drift Time).
- Turn the instrument back to TOF mode in the main Tune page, and thoroughly rinse the system with 50:50 MeOH/H2O before proceeding with the next sample.
4. IMS/IMS-MS processing with MZmine 224
NOTE: MZmine 2 is available from the URL given in the Table of Materials. The use of MZmine 2.51 is recommended. At the time of preparation of this manuscript, the later versions cannot open RAW files from Cyclic IMS instruments because of a change in the import function.
- Import the raw file(s) containing only the IMS and m/z dimensions (*_dt.RAW) using Raw data methods | Raw data import.
NOTE: Raw files will appear on the left side of the main MZmine window. Do not import the original *.RAW files that still contain the retention time dimension. MZmine does not distinguish retention time from IMS arrival time, and the data points of both dimensions would overlap. - Optimize the workflow parameters on a representative file by selecting it in the Raw data files list.
- Evaluate the noise level in the data. Right-click on the file in the Raw data files list, select Show TIC and display the base peak "chromatogram" (BPC). Double-click on the smallest peak observable by eye to display its mass spectrum. Consider the noise level in the data to be around that of the second isotope of the base peak in this spectrum, and use this same value for all the intensity thresholds in the following processing steps.
NOTE: The data were acquired using quadrupole isolation and are thus considered by MZmine as MS/MS. Throughout the entire MZmine processing, be sure to work at an MS level = 2. - Perform the mass detection using Raw data methods | Feature detection | Mass detection. For data acquired in profile mode, use the Wavelet transform algorithm. To set up the parameters of the algorithms in MZmine, click on the […] button next to the algorithm and use the Show preview option to visualize the data while optimizing the parameters.
NOTE: At this stage, the peaks selected by the algorithm will appear in red in the preview window. When using the wavelet transform algorithm on proprietary RAW files, MZmine will sometimes mistake the profile data points for centroided peaks. The software will display a message stating that the user is running a profile algorithm on centroided spectra: ignore this message and click OK. - Reconstruct the extracted ion mobility spectra (EIM) for each fragment mass using Raw data methods | Feature detection | ADAP Chromatogram builder on the 'masses' mass list generated by the previous step. As the m/z tolerance input at this stage is a scan-to-scan tolerance, be sure to leave it at least 3-4 times higher than the overall expected accuracy.
- As the previous step does not have a preview option, check the quality of the peak picking directly using the Feature list that appeared on the right panel of the main MZmine window. Open the Feature list, select all rows, right-click, and select Show/XIC (dialog). Click All to display all ions on the mobility spectrum. Inspect the picked peaks that appear in color to ensure that there are no obvious missed peaks.
- Deconvolve the EIMs to split the m/z that contain different peaks in multiple features. Use Feature list methods | Feature detection | Chromatogram deconvolution, and choose the Wavelets (ADAP) algorithm. Optimize the algorithm for the data using the Show preview option and the following key parameters: S/N threshold, coefficient/area threshold, and RT wavelet range.
NOTE: Checking the aspect of the deconvolved spectrum is recommended. Use the chromatogram visualization tool, as described in step 4.2.4. The deconvolved peaks will appear in color, and peaks of the same mass should be split, as presented in Figure 6A. - Deisotope the deconvolved EIMs using Feature list methods | Isotopes | Isotopic peaks grouper. Use the expected accuracy of the instrument for the m/z tolerance value, and set the arrival time tolerance to 0.1 ms (displayed in MZmine as Retention time tolerance 0.1 min), as isotopes are not resolved during the IMS separation. Check the feature list: if any isotopes remain, increase the tolerance values.
NOTE: Although the deisotoping can theoretically be performed at any moment of the feature list processing, it is important to do it last so that the charge values can be exported (the algorithms used for the other steps will sometimes remove the charge state information).
- Evaluate the noise level in the data. Right-click on the file in the Raw data files list, select Show TIC and display the base peak "chromatogram" (BPC). Double-click on the smallest peak observable by eye to display its mass spectrum. Consider the noise level in the data to be around that of the second isotope of the base peak in this spectrum, and use this same value for all the intensity thresholds in the following processing steps.
- If processing multiple IMS/IMS-MS spectra, repeat the processing with these optimized parameters. Keep the same parameters for all spectra.
- In the case of multiple spectra, group them in a single table to export them; if not, skip directly to step 4.5. To group the spectra, use Feature list methods | Alignment | Join aligner. Because the objective is not to actually align the peaks, use restrictive tolerance values for both m/z and arrival time. Give the same weight to both dimensions.
- Export the final feature list to a *.csv file. Use Feature list methods | Export/Import | Export to CSV file and export the following values: Export row m/z, Export row retention time (the actual IMS arrival time), Peak m/z, and Peak height. Use a comma as a field separator.
5. TWCCSN2 of the centroided IMS/IMS spectra
NOTE: In this protocol, a logarithmic fit calibration25,26 will be used, which tends to give better results than linear calibration and is easy to implement in a spreadsheet or an in-house processing script. An in-house script (written in R) is available at the URL given in the Table of Materials.
- Pick the reference arrival time values from the calibrant acquisition (see step 2.3). Do this manually using the constructor software (see the Table of Materials) to check the aspect of all IMS calibrant peaks.
- In the Chromatogram window, open the *_dt.RAW file corresponding to the calibrant.
- For each calibration point, generate the EIM using the Display | Mass option.
- Check the profile of the EIMs. If some are poorly defined, smooth them using the Process | Smooth option (as the best results are typically obtained with the Savitzky-Golay algorithm, smooth 2 times over 3 bins). Report the apex values in a spreadsheet.
NOTE: Because the reference points are generally acquired using low-resolution DTIMS devices, some multimodal distributions may appear in Cyclic IMS depending on the calibrants. Remove any peak presenting such a distribution from the calibration list.
- Calculate the logarithmic fit parameters from the calibrants.
- For all calibration points, calculate the following.
- Calculate the drift time using Eq (1):
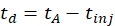 (1)
(1)
with td the drift time, tA the measured arrival time, and tinj the time of injection in the IMS cell (all in ms).
NOTE: For small molecules, such as oligosaccharide fragments, the dead time (flight time between the exit from the IMS cell and the detector) variation between different masses is within the error range of the CCS calibration and can be ignored. - Calculate the neutral mass of the ions using Eq (2):
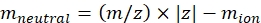 (2)
(2)
with z the charge state of the ion, and mion the mass of the counter-ion (in Da). Use exact masses to avoid introducing uncertainty. If there is an atom loss instead of a counter-ion, use negative mion values (e.g., for [M-H]– mneutral = (m/z) * |z| – (- 1.007276) = (m/z) * |z| + 1.007276). - Calculate the CCS' parameter using Eq (3):
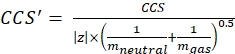 (3)
(3)
with CCS the reference drift tube DTCCSN2 value (in nm2), and mgas the mass of the drift gas (in Da; ex. for nitrogen: mgas = 28.01 Da). - Calculate the td' parameter using Eq (4):
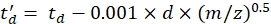 (4)
(4)
with d the detector start delay used experimentally to correct for dead time (typically ~1.5 ms). - Calculate logarithm of the above parameters:
ln (CCS') and ln (t'd)
- Calculate the drift time using Eq (1):
- Perform a linear regression to determine the R2 coefficient and the x and y parameters of the logarithmic fit (with x the slope and ln(y) the intercept) using Eq (5):
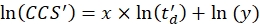 (5)
(5)
NOTE: The user can plot the ln(CCS') vs ln(td') values to visually check the results of the calibration, although this is optional.
- For all calibration points, calculate the following.
- Apply the calibration to the experimental data to calibrate the peaks picked by MZmine for every IMS/IMS spectrum exported to the *.csv file. For each point, calculate the following.
- Calculate the drift time using Eq (6):
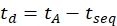 (6)
(6)
with tseq the time preceding the final IMS separation (the 'Time Abs' value noted in step 3.4.6).
NOTE: If calibrating multiple IMS/IMS spectra acquired with different sequences, carefully check the tseq values. - Calculate the neutral mass of the ions using Eq (7):
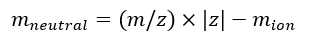 (7)
(7) - Calculate the td' and td'' parameters using Eq (8) and Eq (9):
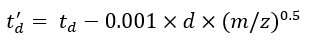 (8)
(8)
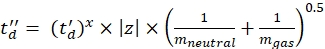 (9)
(9) - Calculate the final calibrated CCS values (TWCCSN2 in nm2) using Eq (10):
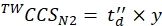 (10)
(10)
NOTE: Although step 5.2.2. gives ln(y) as the intercept, y must be used to obtain the final CCS value. Do not forget to apply an exponential function.
- Calculate the drift time using Eq (6):
- Check the accuracy of the calibration by applying the calibration to the second acquisition of the calibration solution acquired in step 2.4.
NOTE: The calibration should yield results with an error of ~1-2%.
An arabinoxylan pentasaccharide, XA2XX, was chosen as an example to illustrate this protocol. This compound is commercially available, but only as a mixture with another arabinoxylan pentasaccharide, XA3XX (pure XA3XX is also commercially available). The structures of XA2XX and XA3XX are given in Supplemental Figure S1. As the ratio of XA2XX and XA3XX in the commercial mixture is ~50:50, a solution at 20 µg/mL of the mixture was prepared to reach an XA2XX concentration of ~10 µg/mL in 50:50 MeOH/H2O + 500 µM LiCl.
First, an MS analysis of the XA2XX + XA3XX mixture was performed using high-resolution MS. As the two compounds are isomers, a single peak was observed at [M+Li]+m/z 685.24. This MS peak was selected with the quadrupole and the selection window adjusted to remove the -1 Da lithium isotope, which could be mistaken as the monoisotopic peak by processing algorithms (Figure 2).
The [M+Li]+ adducts of the pentasaccharides were then submitted to the first stage of IMS separation: after 3 passes around the cyclic IMS cell, 3 peaks were separated with arrival times of 83, 90, and 94 ms. This profile was compared to that of pure XA3XX (infused at 10 µg/mL), showing that the peaks at 83 and 90 ms corresponded to XA3XX, while the peak at 94 ms corresponded to XA2XX (Figure 4A). The peak at 94 ms was selected for IMS/IMS analysis: the ions belonging to XA3XX were ejected (Figure 4B), and the peak of interest was sent to the prearray store cell. A 3-pass separation was performed after reinjecting the ion without activation to ensure that only the XA2XX peak remained after the selection (arriving at 199 ms in Figure 4C).
Then, the ion was fragmented upon reinjection from the prestore area, and a single-pass IMS separation was performed on all the fragments. Two different activations were tried: the maximum setting of the built-in prestore activation function was first tried (Figure 5B,C); however, the precursor remained the base peak of the spectrum. This is not desired because, for reference spectra, fragments below a certain intensity threshold would typically be removed. Thus, a manually defined prearray gradient → pre-array bias → array offset voltage gradient was chosen (Figure 5D,E).
The generated IMS/IMS-MS data were deconvolved with MZmine 2.51, using the arrival time and m/z dimensions (Figure 6A), to give IMS/IMS spectra containing only the mobility information of the fragments. The peaks above 0.2% relative intensity were exported for CCS calibration (the detailed MZmine parameters are given in Supplemental Table S1). The CCS calibration was performed using the calibration solution (R2 = 0.995, mean absolute deviation of control = 1.63%, see Supplemental Table S3). This processing finally afforded a centroided, CCS-calibrated, IMS/IMS spectrum (Figure 6B).
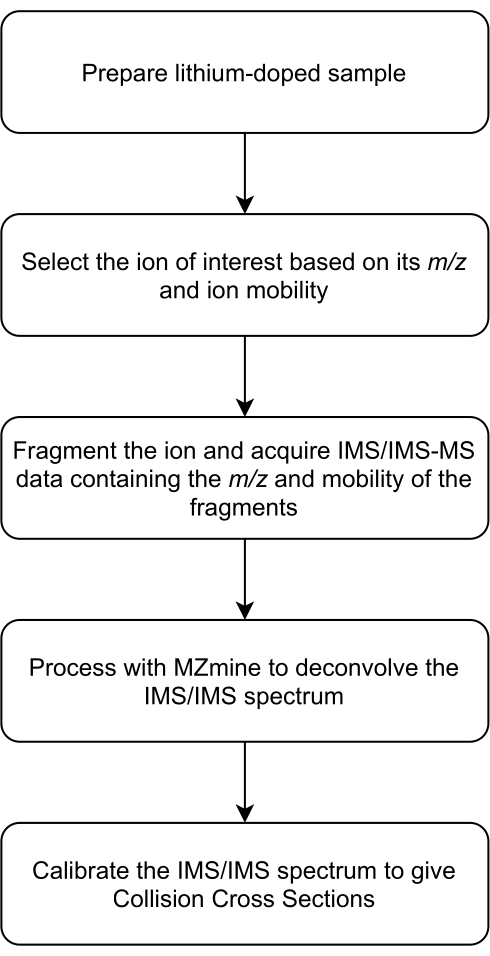
Figure 1: Overview of the IMS/IMS data generation process. Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS. Please click here to view a larger version of this figure.
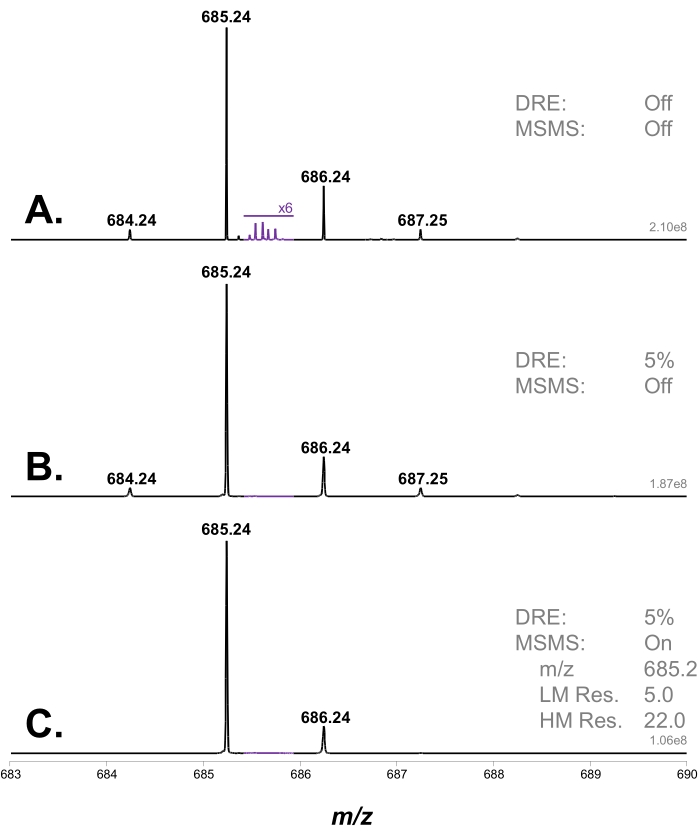
Figure 2: Isotopic pattern of an XA3XX + XA2XX arabinoxylan pentasaccharide mixture. (A) Saturated signal without DRE; (B) signal corrected using DRE with a 5% ion transmission (i.e., 95% attenuation); and (C) profile after quadrupole selection to remove the -1 Da peak corresponding to a lithium isotope. In purple: the region where artifact peaks can appear due to saturation is magnified 6 times. Abbreviations: DRE = dynamic range enhancement; MS = mass spectrometry; MSMS = tandem MS; LM Res = low-mass resolution; HM Res = high-mass resolution. Please click here to view a larger version of this figure.
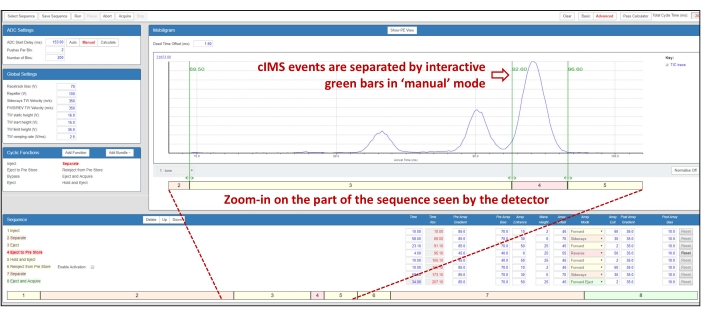
Figure 3: Overview of the Cyclic IMS control window, in which the user defines the IMS/IMS sequence. The sequence displayed shows how to check the quality of the isolation in IMS/IMS, with a selection of XA2XX after 3 passes (the spectrum displayed corresponds to the setting of the selection events after the first-stage separation). The sequence consists in running a first 3-pass IMS separation over 58 ms, then ejecting the two faster isoforms from the IMS cell (segment 3), ejecting the slower isoform (ATD between 92 and 96 ms) in the prestore (segment 4), reinjecting it in the IMS cell without activation (segment 6), allowing the ions to undergo a further 3-pass (58-ms) separation (segment 7), then ejecting ions from the IMS cell, and acquiring data (segment 8). Abbreviations: IMS = ion mobility spectrometry; cIMS = cyclic IMS; IMS/IMS = tandem IMS; ADC = analog-to-digital converter; TW = traveling wave; PE = potential energy; ATD = arrival time distribution. Please click here to view a larger version of this figure.
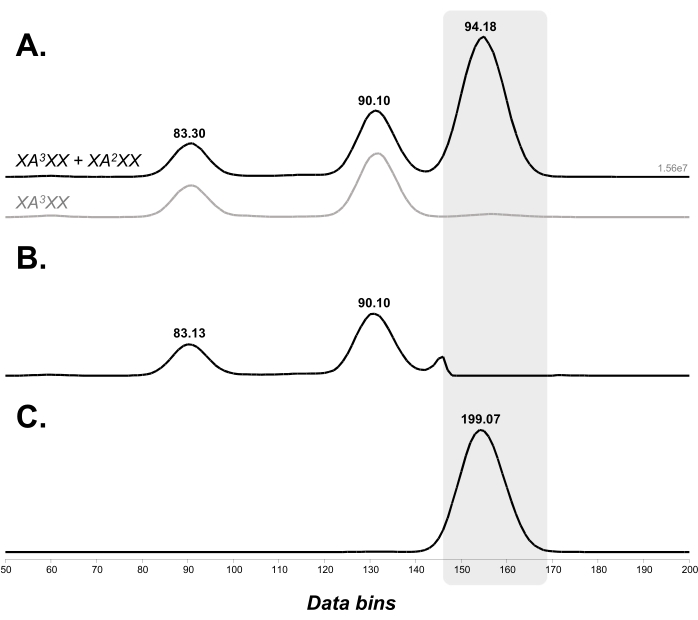
Figure 4: Selection of XA2XX from the mixture of XA2XX and XA3XX. (A) Separation of the arabinoxylan pentasaccharides, XA3XX and XA2XX, after 3 passes (corresponding to a separation time set at 58 ms) around the Cyclic IMS cell. (B) Fraction ejected directly after the first stage of IMS separation. (C) Fraction selected for IMS/IMS on which another 3-pass separation was performed after reinjection. The XA2XX peak of interest is highlighted in gray. The ion mobility spectra are shown in data bins and annotated with their arrival time (ms). Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS. Please click here to view a larger version of this figure.
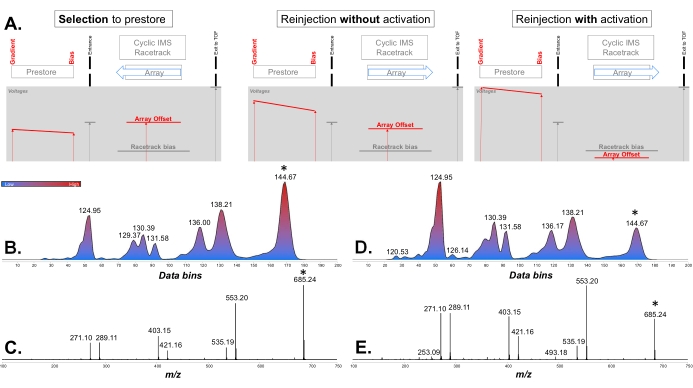
Figure 5: Principles of collision-induced dissociation using the prearray store area. (A) Schematics of the multifunction array region detailing key voltages (in red) used for the selection, the reinjection, and the activation during IMS/IMS experiments. The blue arrows show the direction of the traveling wave in the multifunction array. (B, C) IMS/IMS and MS/MS spectra obtained for XA2XX using the built-in prestore activation function (+150 V). The color bar represents the ion intensity scale (blue = low; red = high). (D, E) IMS/IMS and MS/MS spectra obtained for XA2XX with manual optimization of the voltages (prearray gradient 195 V, prearray bias 180 V, array offset -10 V). Precursor ions are indicated by asterisks on the spectra. The ion mobility spectra are shown in data bins and annotated with their arrival time (ms). Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS; TOF = time-of-flight. Please click here to view a larger version of this figure.
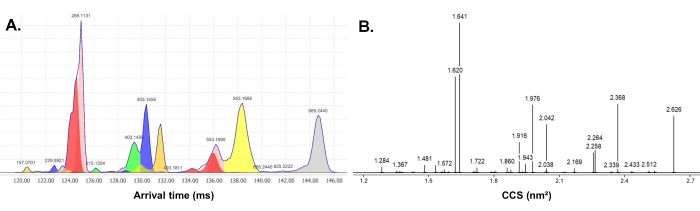
Figure 6: Illustration of the processing steps. Results of (A) the MZmine peak picking and (B) the CCS calibration of arabinoxylan pentasaccharide XA2XX. A shows the mass deconvolution of the IMS/IMS spectrum through a color code. B shows the final IMS/IMS spectrum after centroiding and CCS calibration. Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS; CCS = collision cross section. Please click here to view a larger version of this figure.
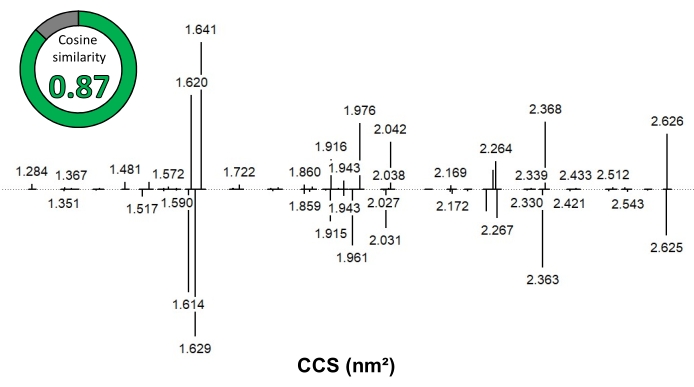
Figure 7: A comparison of two IMS/IMS spectra of XA2XX illustrates the reproducibility of the method. The final calibrated spectrum from this paper (top) is compared to the spectrum from the work by Ollivier et al.21 (bottom, flipped). Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS; CCS = collision cross section. Please click here to view a larger version of this figure.
Supplemental Figure S1: Structures of the XA2XX and XA3XX arabinoxylan pentasaccharides. Please click here to download this File.
Supplemental Figure S2: Evaluation of the interday repeatability using XA2XX. IMS/IMS acquisitions were repeated at Day 1 (top) and Day 95 (bottom). Abbreviations: IMS = ion mobility spectrometry; IMS/IMS = tandem IMS. Please click here to download this File.
Supplemental Table S1: Detailed MZmine parameters. Please click here to download this Table.
Supplemental Table S2: Instrument parameters changed to evaluate the reproducibility. Abbreviation: ESI = electrospray ionization. Please click here to download this Table.
Supplemental Table S3: Control of the CCS calibration using a second acquisition of calibration solution. Please click here to download this Table.
