The stable isotope labeling facilitates the visualization of triglycerides in lipid dynamics studies of female mosquito ovaries. In the 2D mobility domain, the triglyceride 48:1 is displayed in a region with a specific retention time in relation to mobility 1/K0. The intensity of the triglyceride can be visualized by a darker blue line. Other spots represent additional triglycerides found within the ovary. The retention times and mobilities vary due to the differences in masses and structures, therefore appearing in different areas of the graph. These differences are key when distinguishing between triglycerides in a sample (Figure 12A,B). The amplified mass spectrum projection of the region displays a 48:1 triglyceride ion coupled to an ammonium ion at 822 m/z. Other triglycerides, such as 50:2 and 52:3, are also visualized, but at higher m/z regions due to the differences in structure, size, and fragmentation (Figure 12C,D). In this example, we were interested in analyzing the deuterium labelled isotopes. The region for TG 48:1 was further amplified to visualize the isotopically labeled ions. As the amount of deuterium on the ion increases, the intensity of the peak decreases. This happens as there are lower occurrences of the high deuterium labeled ions. The m/z ratio increases slightly with each addition of deuterium due to mass increases. These intensities will further determine the amount of each isotope present within the sample.
When assessing the intensities of the fragments in the mass spectrum, one must compare the non-labeled isotopes with those labeled with 2H (Figure 12E). In blue is the non-labeled intensity with the dashed black lines as theoretical intensities. In red, the isotope labeled results are observed. They display greater intensities than both the non-labeled and theoretical intensities. The enrichment of the species with isotopic labeling yields a greater intensity than the non-labeled theoretical species. The greater intensities allow visualizing the triglycerides due to the district discrepancies. The fragmentation also plays a key role in the identification of lipids, as the PASEF will provide increased fragmentation compared with the full scan MS/MS. This is because the fatty acids in the lipid chains may fragment in different patterns, therefore the lines in the spectra show the intensity of the fragments based on the fragmentation process of the triglyceride (Figure 12F). This provides structural information on the fatty acid and helps to establish which isomer of a species is present. When analyzing data from the DIA method, the number of fragments will increase, therefore resulting in a better MS/MS scan and superior identification than in a typical DDA method.
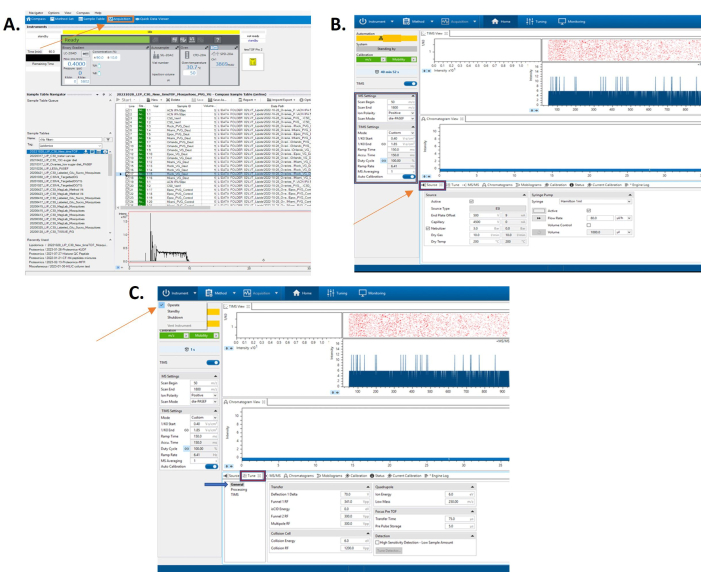
Figure 1: Typical home screen of the software. The home screen allows for method creation and starting the instrument. (A) Software screen containing TIMS and MS/MS control settings. At the bottom of the screen is the source tab to edit those controls on the instrument. (B) Software screen with the tune tab opened. (C) Here, one can prepare the instrument for general tune. Please click here to view a larger version of this figure.
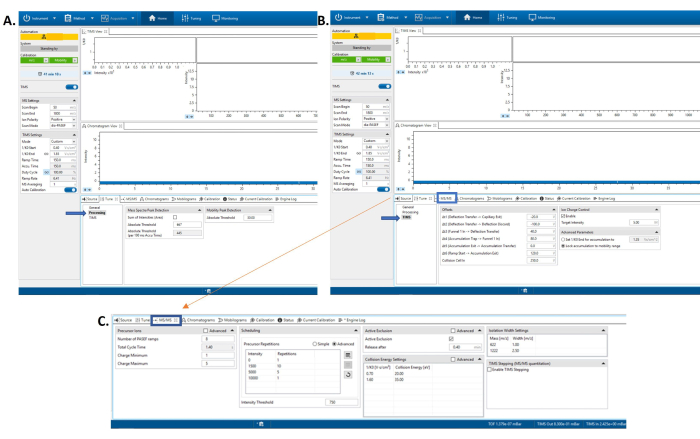
Figure 2: Software screen with the tune tab opened. Here one can edit the processing tune parameters. (A) Software screen with the tune tab opened. Here one can edit the tune parameters for the TIMS portion of the instrument. (B) Software screen with the MS/MS tab selected at the bottom of the screen. (C) Here one can edit parameters for the tandem mass spectrometer. Please click here to view a larger version of this figure.
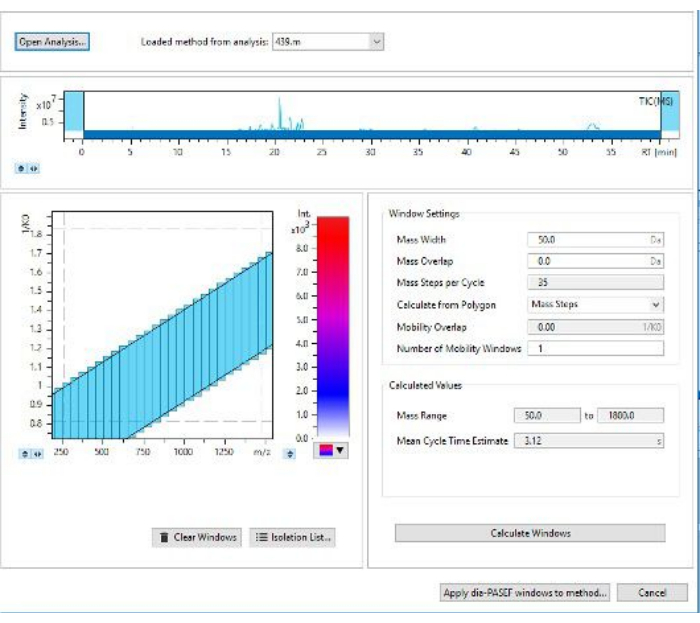
Figure 3: Screen for selecting DIA-PASEF window widths. Here one can edit the colors for abundance as well as edit the size of the windows for data acquisition. Please click here to view a larger version of this figure.
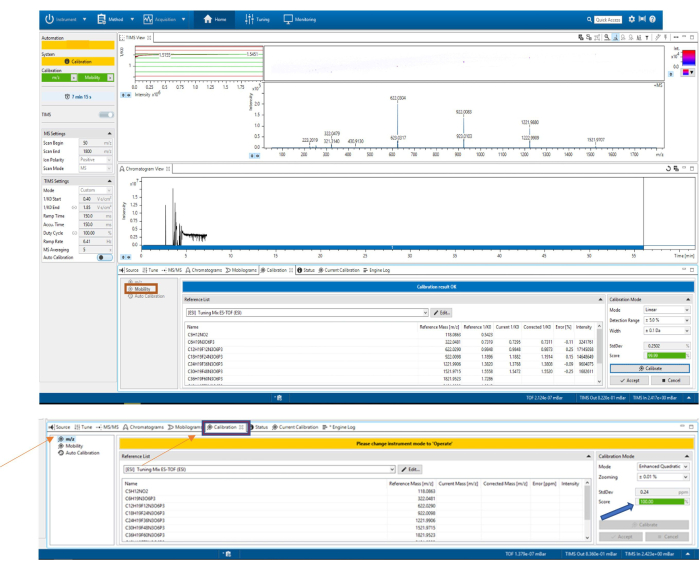
Figure 4: Calibration of the mass spectrometer by mass to charge ratio (m/z). The reference list is selected for the calibration. (A) At the bottom right, the Calibrate button is clicked until an acceptable value appears. Calibration of the ion mobility spectrometer by mobility. The reference list is selected for the calibration. (B) At the bottom right, the Calibrate button is clicked until an acceptable value appears. Please click here to view a larger version of this figure.
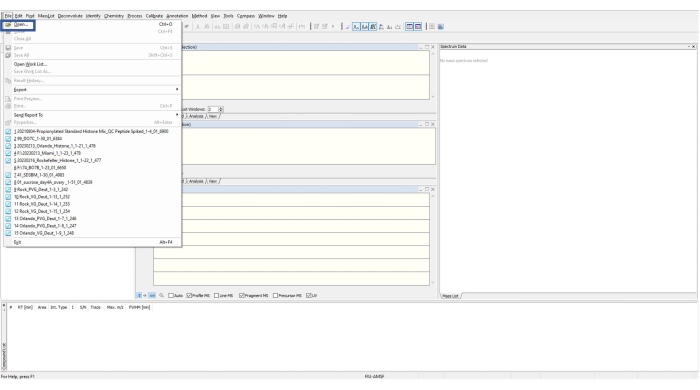
Figure 5: Typical home screen for the data analysis software. To open a file, the top left File button is clicked for the drop box with options to appear. Please click here to view a larger version of this figure.
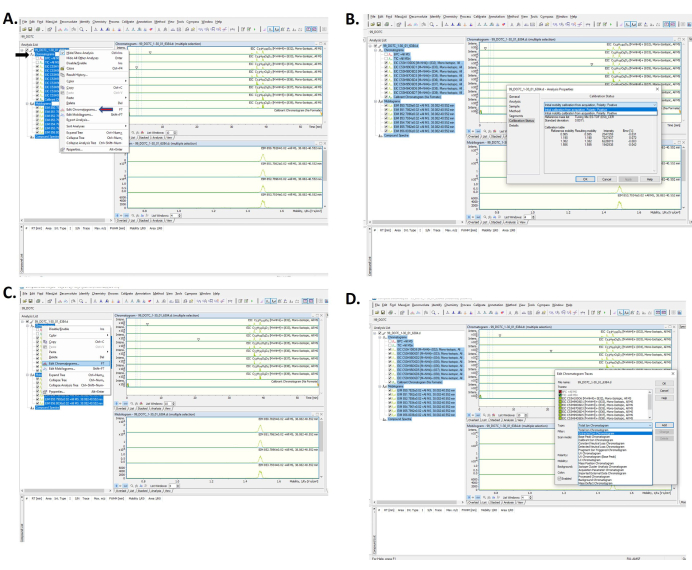
Figure 6: Data analysis software. The software screen confirming a successful calibration with (A) how to select calibration results and (B) selecting the calibration category. (C) Selecting a chromatogram to analyze and (D) selecting an extracted ion chromatogram. Please click here to view a larger version of this figure.
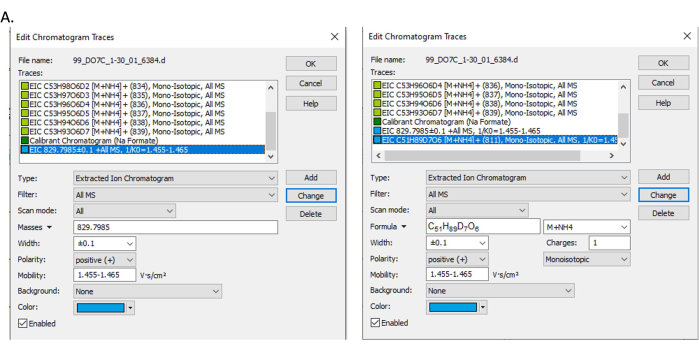
Figure 7: Pop-up screen. Pop-up for selecting and extracting ion chromatogram by (A) mass filter and (B) formula filter. Please click here to view a larger version of this figure.
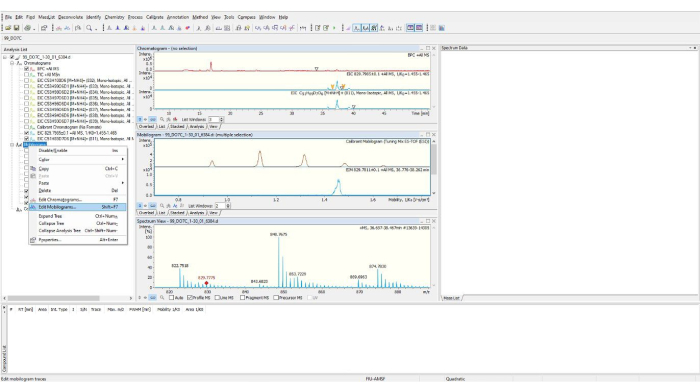
Figure 8: Data Analysis software. The software screen where at the left, a mobilogram is selected for analysis. Please click here to view a larger version of this figure.
Figure 9: Pop-up screen for mobilogram. Pop-up for selecting an extracted ion mobilogram by (A) mass filter and (B) formula filter. Please click here to view a larger version of this figure.
Figure 10: Data analysis screen. (A) The software screen; highlighted on the left one can form an extracted ion list. (B) Data analysis screen where at the bottom a compound spectrum from the extracted ions is prepared. Please click here to view a larger version of this figure.
Figure 11: Data analysis screen. The software screen demonstrating the results of a manual integration of compound spectra. Please click here to view a larger version of this figure.
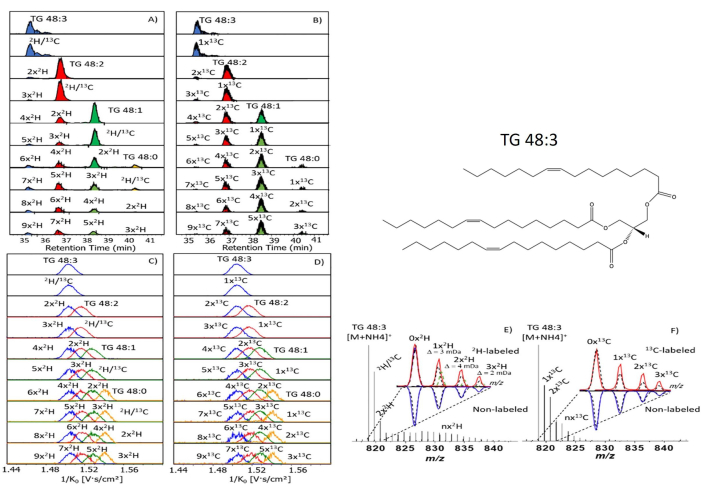
Figure 12: LC-TIMS MS Result. (A) The LC-TIMS MS at 4 days after feeding 2H-labelled sucrose diet and the insect ovaries can be seen in the 2D mobility domain. (B) Typical MS projections for the signal corresponding to TG 48:1, TG 50:2 and TG 52:3 from labelled sample. (C) Amplified MS region 822 – 827 m/z denoting labelled TGs profile. (D) Pay particular attention to MS peaks in red are amplified the isotope profile. (E-F) The 2H labelled isotope profile is highlighted in red; below non-labelled isotope profile are highlighted in blue. The theoretical isotope profiles are indicated as dash lines (13C in black; 2H in green). A potential structure for triglyceride 48:3 is also displayed on the top right. Please click here to view a larger version of this figure.
