Intermuscular adipose tissue (IMAT) is an ectopic adipose depot residing between and around muscle fibers1. As described in detail in a recent review by Goodpaster et al., IMAT can be detected using high-resolution computed tomography (CT) and magnetic resonance imaging (MRI) (Figure 1A,B) and is found around and within muscle fibers throughout the entire body1. The quantity of IMAT varies greatly between individuals and is influenced by BMI, age, sex, race, and sedentariness2,3,4. Moreover, IMAT deposition is commonly seen in pathological conditions associated with muscle degeneration5, and numerous studies have documented increased IMAT mass in individuals with obesity, type 2 diabetes, metabolic syndrome, and insulin resistance6,7,8,9. Nonetheless, the cellular and biological properties of IMAT are only beginning to be unraveled. The limited accessibility and the variation in IMAT locations and content throughout the body have challenged the collection of samples from this unique adipose depot2. Moreover, samples are easily 'contaminated' with skeletal muscle (SM) upon collection, making the separation between the biological contribution from the different tissues difficult to decipher (Figure 1C). To this end, single nuclei RNA sequencing (snRNA-seq), which has gained considerable attention during the last decade, serves as an ideal methodology to allow for the separation of IMAT- and SM-derived gene expression patterns with single-cell resolution. Moreover, nuclei isolation is particularly useful for adipose tissue due to the large lipid-laden adipocytes, which are impossible to dissociate into single-cell suspension without compromising the integrity of the cells. Lastly, this technology holds the potential to discover novel markers of IMAT-specific adipocytes and uncover the composition and presence of different progenitor cell populations, as well as study the variation of the cell composition in pathological and normal conditions.
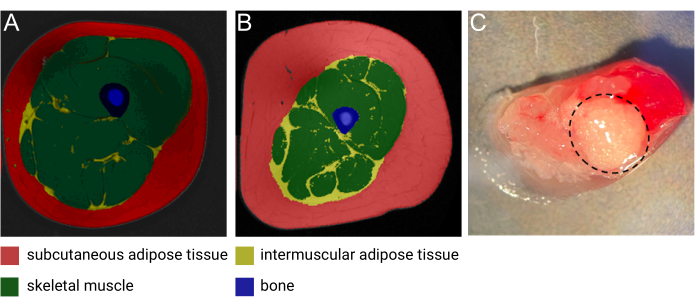
Figure 1: Images of IMAT. Representative magnetic resonance (MRI) image of IMAT from (A) a middle-aged lean female and (B) a middle-aged male with obesity. Red: subcutaneous adipose tissue, yellow: intermuscular adipose tissue, green: skeletal muscle, blue: bone. Image courtesy of Heather Cornnell, AdventHealth Translational Research Institute. (C) Fresh tissue sample with IMAT (encircled by dashed black line). Image courtesy of Meghan Hopf, AdventHealth Translational Research Institute and Bryan Bergman, University of Colorado. This figure has been modified with permission from Goodpaster et al.1. Please click here to view a larger version of this figure.
A number of studies have been published from the livestock industry investigating the marbling of meat (IMAT in particular) in pigs, chickens, and cattle using single-cell (sc) and snRNA-seq10. These studies have identified several subpopulations of adipocytes and markers of potential progenitor cells of IMAT11,12,13; however, whether these cellular compositions translate to human IMAT is unknown. To our knowledge, only one study has looked into the cellular heterogeneity of human muscle with fatty infiltration, obtained from male patients with hip osteoarthritis, using snRNA-seq14. The investigators reported a small adipocyte population and several fibro-adipogenic progenitor (FAP) subpopulations within the large population of myonuclei14. Our study is the first to develop a method to directly interrogate IMAT manually dissected from human muscle for cellular composition using snRNA-seq.
Importantly, protocols for snRNA-seq need to be customized for the specific tissue studied, as the amount of tissue available and the physical properties of the specific tissue will dictate the optimal processing steps. The tissue yield for IMAT is typically small, often not exceeding 50 mg, even when performing ultrasound-guided biopsies. Hence, proper processing of this scarce tissue is essential. We believe that this protocol will serve as a valuable resource for researchers studying human IMAT.
The sample used for this protocol was part of the Study of Muscle, Mobility, and Aging (SOMMA)15, which was approved by the Western IRB-Copernicus Group (WCG) Institutional Review Board and was carried out in accordance with the Declaration of Helsinki. Participants provided written informed consent for their participation in the study.
NOTE : This protocol is adapted from a previous protocol using 100 mg of human abdominal subcutaneous adipose tissue on a nanowell-based platform16. The current protocol is optimized for 50 mg of human IMAT and library preparation using a droplet-based platform. Further optimization of this protocol for nuclei isolations from non-human IMAT or other adipose depots may be required.
1. Preparation of buffers and reagents (Table 1 and Table 2)
NOTE: Prepare buffers fresh on the day of the experiment and do not re-use.
- Pre-cool a centrifuge to 4 °C.
- Prepare homogenization buffer and nuclei isolation medium.
- Obtain two buckets of ice and pre-cool 2 x 15 mL conical tubes.
- Mix all reagents for the homogenization buffer (HB) in a 15 mL conical tube in the order listed in Table 1. Keep on ice. Mix by vortexing.
- Mix all reagents for nuclei isolation medium (NIM) in a 15 mL conical tube in the ordered list in Table 2. Keep on ice. Mix by vortexing.
- Prepare 10% Triton-X by adding 100 µL of Triton X-100 to 900 µL of Nuclease-free water. Vortex to ensure proper mixing. Keep at room temperature (RT).
| Reagent | Volume (μL) | Final concentration (mM) | |
| 1x | 2x | ||
| 1 M MgCl2 | 10 | 20 | 5 |
| 1 M Tris Buffer, pH 8.0 | 20 | 40 | 10 |
| 2 M KCl | 25 | 50 | 25 |
| 1.5 M Sucrose (-4oC) | 334 | 668 | 250 |
| 1mM DTT | 2 | 4 | 0.001 (~1 µM) |
| 100x protease inhibitor | 20 | 40 | 1x |
| Superasin 20 U/µL | 40 | 80 | 0.4 U/µL |
| Nuclease-free water | 1549 | 3098 | – |
| Total Volume | 2000 | 4000 | – |
Table 1: Homogenization buffer (HB). Keep on ice. Mix by vortexing.
| Reagent | Volume (μL) | Final concentration (mM) | |
| 1x | 2x | ||
| EDTA | 0.4 | 0.8 | 0.1 |
| Ribolock RNAse inhibitor (40U/µL) | 40 | 80 | 0.8 U/µL |
| 1% BSA-PBS (-/-) | 1959.6 | 3919.2 | – |
| Total Volume | 2000 | 4000 | – |
Table 2: Nuclei isolation medium (NIM). Keep on ice. Mix by vortexing.
2. Pulverization of frozen tissue ( Figure 2A)
- Set up the workstation for homogenization.
- Fill up the cannister with liquid nitrogen (LN2).
CAUTION: when working with LN2, always wear goggles and cryo gloves. - Obtain 2x mortars, 1x pestle, 1x micro-scoop spatula, 1x glass dounce, and 1x stainless-steel pestle for the automatic douncer.
- Set up the automatic douncer.
- Fill a beaker with ice and pre-cool the glass dounce.
- Fill up the cannister with liquid nitrogen (LN2).
- Fill the 2 mortars (containing the pestle and spatula) with LN2 to cool down the instruments. Let the LN2 evaporate and repeat.
- While instruments are cooling, add 1 mL of HB to the glass dounce.
- Fill both mortars with LN2 one last time and pour the 50 mg IMAT sample into one of the mortars.
- Pulverize the IMAT using the pestle by gently pressing it down on the piece of tissue to break it into small pieces. Make sure all pieces are pulverized.
- The LN2 will slowly evaporate while pulverizing the tissue. When tissue is properly pulverized, and there is still ¼ – ½ of a mortar of LN2 left, tilt the mortar towards the lip of the mortar to collect the pulverized tissue by the lip. Let the LN2 evaporate completely.
- Immediately after the last LN2 has evaporated, scoop the pulverized tissue into the glass dounce containing 1 mL of HB.
3. Homogenization of pulverized tissue
- Homogenize the pulverized tissue using the automatic douncer. Bring the glass dounce up and down the stainless-steel pestle for 10 strokes in the forward direction, followed by 10 strokes in the reverse direction.
- Ensure that the solution is cloudy after homogenization and contains no visible pieces of tissue. A light pink color is often expected due to contamination with muscle tissue.
- Transfer the homogenate to a pre-cooled 1.7 mL low-bind tube on ice.
- Use 400 µL of HB to rinse the dounce to make sure all the material is transferred and add it to the tube.
NOTE: Two samples can be processed at a time. To do this, double the amount of HB and NIM. Pulverize and homogenize one tissue sample and immediately thereafter pulverize and homogenize the second tissue sample to be able to perform the isolation and clean-up steps in parallel.
4. Nuclei isolation and clean-up ( Figure 2B)
- Add 14 µL of Triton-X (10%) to the homogenate for a 0.1% concentration.
- Keep the tube on ice and in the dark for 10-15 min while vortexing every 3 min.
- Pre-wet one 100 µm and one 40 µm cell strainer (per sample) with 100 µL of RT DPBS for each in a 50 mL conical tube.
- Filter the homogenate through the 100 µm cell strainer.
- Rinse the 1.7 mL tube with 400 µL of HB and filter through the 100 µm cell strainer.
- Next, filter the solution through the 40 µm cell strainer.
- Transfer an equal amount of solution into two pre-cooled 1.7 mL low-bind tubes corresponding to ~900 µL in each tube.
- Centrifuge the tubes for 10 min at 2700 x g at 4 °C. There should be a small pellet visible after centrifugation.
- Remove and discard the top lipid layer and the remaining supernatant, leaving ~50 µL solution from the first tube.
- Repeat for the second tube.
- Thoroughly resuspend the pellet in the first tube by gently pipetting up and down 20x and transfer to a new 1.7 mL low-bind tube. Avoid creating bubbles.
- Repeat this procedure for the second tube and transfer the resuspended solution to the same tube.
- Add 500 µL of NIM and mix with pipetting.
- Centrifuge the tube with a balance at 1000 x g for 10 min at 4 °C.
- Remove supernatant, leaving ~50 µL, and gently pipette up and down until the pellet is resuspended. Optionally, transfer the resuspended pellet to a new clean tube if some lipid leftover remains on the side of the tube.
- Add 200 µL of NIM and mix by pipetting.
5. Nuclei staining and counting (Figure 2C and Figure 3)
NOTE : To facilitate the counting, set up a 'nuclei counting' protocol on an automated cell counter, as adjusting the bright field and DAPI channels can affect the count greatly. Adjust the channels so that only nuclei and not debris are captured. Make sure the bright field channel only marks 'objects' that also have a DAPI stain.
- Add 1 drop of the live cell staining solution and leave it in the dark, on ice, for 15 min.
- Filter the solution through a 30 µm cell strainer.
- Mix the nuclei solution by pipetting and add 10 µL of the solution to a cell counting chamber slide.
- Count nuclei using an automated cell counter.
NOTE: The optimal concentration is 1000 nuclei/µL corresponding to 1.0 x 106/mL.- Ensure no clumps of nuclei are present, as this could clog the chip for generating single nuclei droplets (Figure 3).
- If the nuclei concentration is not high enough, spin down the solution at 1000 x g for 10 min at 4 °C to gain a pellet, remove the supernatant, and resuspend in a smaller volume.
- If the degree of debris in the solution is high, resuspend the nuclei solution in a larger volume of NIM (i.e., 1 mL) and filter through a 30 µm cell strainer again. Then spin down at 1000 x g for 10 min at 4 °C and resuspend in appropriate volume in relation to the concentration of nuclei.
- After obtaining the nuclei concentration, proceed directly to the first step in the library preparation.
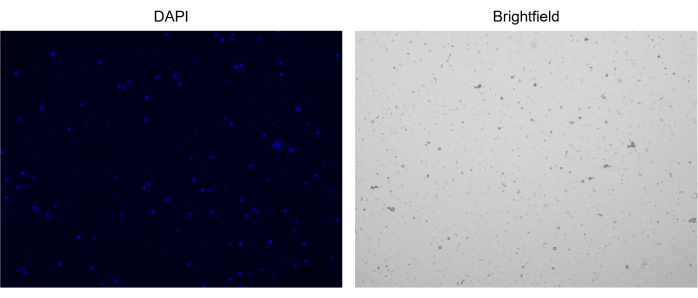
Figure 3: Staining of isolated nuclei. Image from cell counter of nuclei stained with NucBlue/DAPI (left image) and the corresponding bright field image (right image). The presence of minor amounts of debris is evident in the bright field image. The automated cell counter used here does not have an option to include scale bars. Please click here to view a larger version of this figure.
6. Library preparation and sequencing parameters
- Refer to a thorough protocol for library preparation using the droplet-based single nuclei approach available on the provider's webpage17.
- Aim for a targeted nuclei recovery of 10,000. However, for samples with a high level of debris or fragile nuclei, a lower number of nuclei recovered would be anticipated.
- Store the samples at 4°C for up to 72 h after step 2.3. in the library preparation protocol to combine the processing of more samples in parallel. Do this by processing two samples until step 2.3 on two consecutive days, and on the third day, process the 4 samples together from step 3 and forward in the library preparation protocol.
- Sequencing parameters: Sequence on a sequencing platform aiming for 50,000 paired-end reads per nuclei.
NOTE: The data presented in this protocol was sequenced on the NovaSeq 6000 platform, aiming for 50,000 paired-end reads per nuclei.
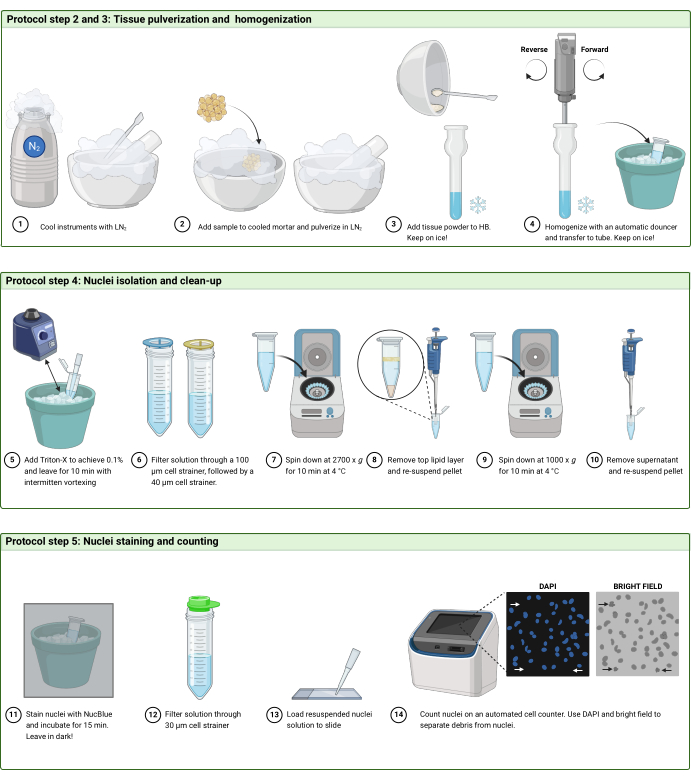
Figure 2: Protocol workflow. Schematic illustration of the workflow in (A) steps 2 and 3, (B) step 4, and (C) step 5 of the protocol. The figure was created with BioRender.com. Please click here to view a larger version of this figure.
7. Data processing and analysis
NOTE : In this protocol, some of the recommended software and R packages used to process the resulting sequencing data are briefly introduced, focusing on the steps after the initial pre-processing (Table 3). This study provides general quality control (QC) metrics and an example uniform manifold approximation and projection (UMAP) in Figure 4. However, an in-depth description of the bioinformatic analysis is out of the scope of this protocol. Therefore, readers can refer to the recent review on best practices for single-cell analysis by Heumos et al.18.
- Pre-processing of sequencing data
- Map the single nuclei reads to the human reference genome GRCh38.
- Include intron reads in the count.
- Perform QC and filtering of the data using the Seurat R package19.
- Compute a cell complexity score by dividing the log(10) number of genes detected by the log(10) number of reads detected.
- Plot out the most important QC metrics using a histogram or violin plot, including the number of genes detected per nuclei, mitochondrial read percentage, and cell complexity score.
- Filter out nuclei with less than 200 or more than 10,000 genes per nuclei, greater than 10% mitochondrial reads, and a complexity score below 0.8.
- Normalize data and perform dimensionality reduction.
- Use the SCTransform function from Seurat to normalize the data using 2000 variable features.
- Cluster data using the following functions from the Seurat R package: RunPCA, FindNeighbors, FindClusters, and RunUMAP.
- Plot a UMAP to visualize the clustering of the data.
- Filter out predicted doublets using the DoubletFinder R package20 and re-cluster data.
- Annotate clusters using known gene markers of the cell types expected to be present in the tissue (supervised approach) or based on the top 5 differentially expressed genes between the clusters (unsupervised approach).
- Use decontX21 to determine the degree of ambient RNA contamination and to adjust the gene expression matrix for ambient RNA.
- Include the raw gene matrix as background.
- Save the Seurat object for future exploration of the data.
NOTE: The code for QC and clustering analysis is available in Supplementary File 1.
| Software/R packages used in data workflow | Alternative software/packages | Processing step |
| CellRanger | STARsolo, kallisto | Trimming, alignment, mapping |
| Seurat | SingleCellExperiment, Cellranger | QC, analysis and data exploration |
| DoubletFinder | scds, scdblFinder, Scrublet | Doublet detection |
| DecontX | SoupX, CellBender | Ambient RNA adjustment |
Table 3: Software/tools for data workflow.
This workflow was designed to guide the processing of frozen human IMAT samples to obtain gene expression profiles at single nuclei resolution, enabling cell type identification. Here, one representative IMAT sample from a participant in the SOMMA study is presented.
The first step of any analysis of snRNA-seq data is to evaluate the quality of the data to identify poor-quality nuclei, which should potentially be removed from the dataset. Importantly, the filtering steps and thresholds should be determined for the specific type of sample and dataset you have in hand, as the commonly evaluated metrics can differ among tissues and cell types22,23. Figure 4A provides visuals of some of the key metrics used to assess the quality of the generated snRNA-seq data. The number of genes detected per nuclei is dependent on sequencing depth and cell type but would be expected to be above 200 for good-quality nuclei18,23. It was found that the data generated using this protocol is within the expected range with a median of 1134 genes per nuclei, from a total of 4662 nuclei.
The percentage of mitochondrial reads is evaluated since a high degree of mitochondrial contamination can arise from damaged nuclei or ambient RNA attaching to the nuclei, indicating poor-quality nuclei. In the dataset presented here, a median mitochondrial read percentage of 2.65 was found, which is well below the 5%-20% threshold commonly used in the literature24,25,26. The percentage of ribosomal reads differs among cell types and tissues. However, as large proportions of ribosomal genes can influence the clustering of the data, it is recommended to check the ribosomal read percentage and potentially remove ribosomal genes or nuclei with high levels of ribosomal genes from the dataset before clustering. The data generated with this protocol showed a low level of ribosomal reads with a median of 2.46% and a maximum of 16.5%, and therefore, we did not filter based on this metric. Lastly, a cell complexity score was calculated based on the log(10) number of genes detected divided by the log(10) number of reads detected. Good-quality nuclei are expected to be above 0.8, and a median of 0.92 was obtained in the sample used in this study. Based on these QC metrics, one can decide which nuclei to filter out of the dataset. For analysis, we chose to filter out nuclei with less than 200 or more than 10,000 genes per nuclei, greater than 10% mitochondrial reads, and a complexity score below 0.8.
Following the initial quality assessment and filtering step, a UMAP can be generated to visualize clustering of the nuclei. Clustering was performed based on the 2000 most variable genes using SCT transformation. The initial clustering steps can be used to check if any of the QC features cluster together, e.g., nuclei with high mitochondrial reads. Moreover, clustering information is required for some doublet detection methods, including DoubletFinder20, which was used in this protocol. DoubletFinder was used with an expected multiplet rate set to 4.8% as suggested by the providers of the droplet-based platform. After doublet removal, the level of ambient RNA contamination was estimated, which is particularly common in single nuclei preparations as RNA is released from the cytoplasm upon cell lysis and gets dispensed into the Gel Beads-in-emulsion (GEMs) and amplified in the following library preparation steps. Hence, several tools have been developed to correct the inherent problem of ambient RNA contamination (see Table 3). We used the R package decontX21, in which the raw background matrix (including only empty droplets) is used to adjust the gene expression matrix, enhancing the real gene expression signature.
The clustering and ability to detect low abundant cell types depend on the number of nuclei. This study detected all expected major cell types in IMAT (Figure 4B) from a total of 3817 nuclei after QC filtering, doublet removal, and ambient RNA adjustment. These included stem cells, fibro-adipogenic progenitors (FAPs), and mature adipocytes, as well as pericytes, smooth muscle cells, immune cells, muscle progenitor cells, and myonuclei from skeletal muscle cell contamination.
Overall, we have demonstrated that this protocol produces high-resolution single nuclei data enabling detection of cell type annotation important for unravelling the biology and cellular origins of IMAT.
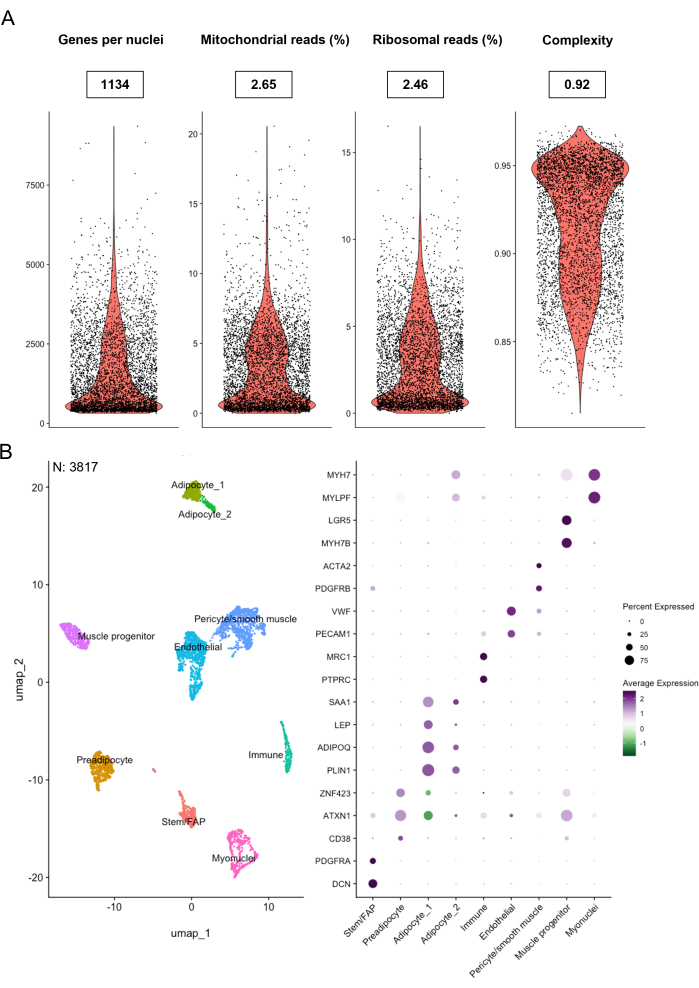
Figure 4: Quality assessment, clustering, and cell type annotation of sequencing data. (A) Violin plots of essential metrics for evaluation of the sample and sequencing performance, including the number of genes detected per nuclei, percentage of mitochondrial reads, percentage of ribosomal reads, and cell complexity measured as the log(10) number of genes detected divided by the log(10) number of reads detected. Median values for each metric are given in closed boxes. Total number of nuclei: 4662. (B) UMAP displaying clustering of individual nuclei and corresponding DotPlot showing relative gene expression of cell type marker genes for each cluster after filtering. Number of nuclei: 3817. Please click here to view a larger version of this figure.
Supplementary File 1: The code for QC and clustering analysis. Please click here to download this File.
